BioPROTAC: A Complementary Approach of PROTAC for Protein Degradation
Consult with Our Experts
Article
What Is BioPROTAC?
BioPROTAC, also written as biological PROTAC, is a protein-based targeted degradation strategy designed to remove a protein of interest through the ubiquitin-proteasome system. While a conventional PROTAC is typically a chemically synthesized heterobifunctional molecule composed of a target ligand, a linker, and an E3 ligase ligand, a BioPROTAC uses engineered biological recognition elements to bring a target protein into proximity with ubiquitination machinery. In a typical BioPROTAC format, one module recognizes the protein of interest, while another module contains or recruits E3 ubiquitin ligase activity. Once these modules place the target near a productive ubiquitination environment, the target can be polyubiquitinated and directed to the proteasome for degradation.
The concept is especially attractive for research programs in which small-molecule ligand discovery is difficult. Many proteins involved in signaling, transcriptional control, scaffolding, protein-protein interaction networks, or protein aggregation do not present a deep catalytic pocket suitable for classical small-molecule inhibition. BioPROTAC design can instead use peptide binders, nanobodies, monobodies, engineered protein binders, or other high-affinity biological recognition domains. This allows researchers to test whether a target is degradable even before a chemical warhead is available, and it can provide a fast route to target validation at the protein level.
BioPROTAC should therefore be viewed as a complementary approach rather than a replacement for small-molecule PROTACs. Small-molecule PROTACs remain highly valuable when target ligands, E3 ligase ligands, linker chemistry, cellular permeability, and structure-activity relationship workflows are well established. BioPROTACs add another layer to targeted protein degradation by expanding the types of binders, E3 modules, delivery formats, and biological control strategies that can be tested in research systems.
Definition of BioPROTAC in Targeted Protein Degradation
A BioPROTAC is generally an engineered protein or biologically encoded degrader that combines a target-recognition domain with an E3 ligase-related degradation module. The target-recognition domain may bind a native protein, a tagged protein, a specific conformation, or a post-translationally modified state. The degradation module may include a functional E3 ligase domain, an E3 adaptor-binding domain, or another element capable of positioning the target for ubiquitin transfer. The design goal is not simply binding, but productive proximity: the BioPROTAC must place lysine residues or other ubiquitination-accessible regions of the target in a geometry that supports ubiquitin chain formation and subsequent proteasomal turnover.
Why BioPROTAC Emerged as a Complementary Strategy to PROTAC?
Small-molecule PROTAC discovery can be slowed by the need for high-quality target ligands, suitable attachment sites, linker optimization, E3 recruiter selection, and cellular exposure. BioPROTAC design can bypass some of these early bottlenecks by using already available protein binders or rapidly generated affinity reagents. For example, a nanobody against a target surface may be fused to an E3-related domain and expressed in cells to evaluate whether target degradation is feasible. If degradation is observed, researchers gain evidence that the target can be removed through induced proximity and can then decide whether to pursue small-molecule PROTACs, molecular glues, BioPROTACs, or other degradation modalities.
Key Components of a BioPROTAC Molecule
The modular design of BioPROTACs makes them flexible, but each module must be selected with the overall degradation mechanism in mind. A high-affinity binder alone is not sufficient if the target epitope is poorly positioned for ubiquitination. Likewise, an active E3 module may not produce degradation if the fusion orientation or linker geometry prevents productive contact with the target protein.
Table 1. Core Design Elements of a BioPROTAC.
| Component | Function in BioPROTAC Design | Key Optimization Questions |
|---|
| Target-binding module | Recognizes the protein of interest through a peptide, nanobody, monobody, designed protein binder, or other biological binder. | Is the epitope accessible? Is the binder selective? Does binding preserve the desired degradation geometry? |
| E3 ligase-related module | Provides or recruits ubiquitination activity to the target protein. | Is the E3 active in the selected cellular context? Is its localization compatible with the target? |
| Linker or fusion architecture | Controls distance, orientation, flexibility, and steric accessibility between modules. | Should the binder be placed at the N-terminus or C-terminus? How long and flexible should the linker be? |
| Expression or delivery strategy | Places the BioPROTAC inside the relevant cellular compartment. | Will the degrader be genetically encoded, delivered as a recombinant protein, or introduced through another research format? |
| Assay readouts | Confirms target engagement, ubiquitination, degradation, selectivity, and functional response. | Which assays distinguish true degradation from reduced expression, altered localization, or assay interference? |
How Does BioPROTAC Work?
BioPROTACs operate through induced proximity. Instead of inhibiting a single enzymatic activity, they are designed to change the fate of the entire target protein. The target-binding module captures the protein of interest, the E3-related module promotes ubiquitin transfer, and the proteasome recognizes the ubiquitinated target for degradation. This mechanism is especially useful for research questions where protein abundance, scaffolding function, protein complex formation, or non-enzymatic activity is more relevant than catalytic inhibition.
Target Recognition by Protein-Based Binding Domains
The first step is selective recognition of the target protein. BioPROTACs may use a peptide sequence that binds a defined motif, a nanobody that recognizes a folded surface, a monobody that binds a protein interaction interface, or an engineered binder selected by display or computational design. Compared with chemical ligands, protein binders can recognize broader surfaces and can be optimized for conformational or isoform selectivity. This is one reason BioPROTACs are frequently discussed for proteins that lack classical binding pockets. Target recognition must also be evaluated in the cellular environment. A binder that performs well in purified protein assays may fail inside cells if the epitope is masked by a partner protein, the target resides in a different compartment, or the binder changes target localization. Therefore, early BioPROTAC development usually benefits from parallel biochemical, cellular, and imaging-based assays.
E3 Ligase Recruitment and Ubiquitin Transfer
The second step is recruiting or providing E3 ubiquitin ligase activity. In many BioPROTAC designs, an E3 ligase domain or E3 adaptor-related domain is fused directly to the target-binding module. This differs from small-molecule PROTACs, in which a chemical E3 ligand recruits an endogenous E3 complex. Direct fusion offers flexibility because researchers can test E3 domains that may not yet have small-molecule recruiters. However, it also adds design considerations related to protein folding, activity, localization, and expression level. Successful ubiquitin transfer depends on more than proximity. The degrader must orient the target so that accessible residues can receive ubiquitin chains, and the E3-related module must remain catalytically competent. Controls such as inactive E3 mutants, non-binding target modules, proteasome inhibition, and ubiquitination assays are important for confirming that the observed decrease in target abundance is caused by the intended ubiquitin-proteasome mechanism.
Proteasome-Mediated Degradation of the Target Protein
After ubiquitination, the target protein is recognized by proteasomal machinery and degraded. The measurable outcome is a reduction in target protein level, often quantified by Western blotting, ELISA, flow cytometry, cellular imaging, or proteomic analysis. Key parameters include degradation depth, degradation kinetics, time to maximum degradation, recovery after washout or expression shutoff, and selectivity against related proteins. For BioPROTACs, interpretation should consider the expression level of the degrader itself, because excessive expression can change stoichiometry, localization, or cellular protein quality-control behavior.
BioPROTAC vs. Small-Molecule PROTAC: Similarities and Differences
BioPROTACs and small-molecule PROTACs share the same conceptual foundation: a target protein is brought into proximity with an E3 ubiquitin ligase to promote ubiquitination and degradation. The practical workflow, however, can be quite different. Small-molecule PROTACs are built through chemical synthesis and optimized through medicinal chemistry. BioPROTACs are built through protein engineering, binder selection, fusion architecture optimization, expression control, and intracellular delivery strategy.
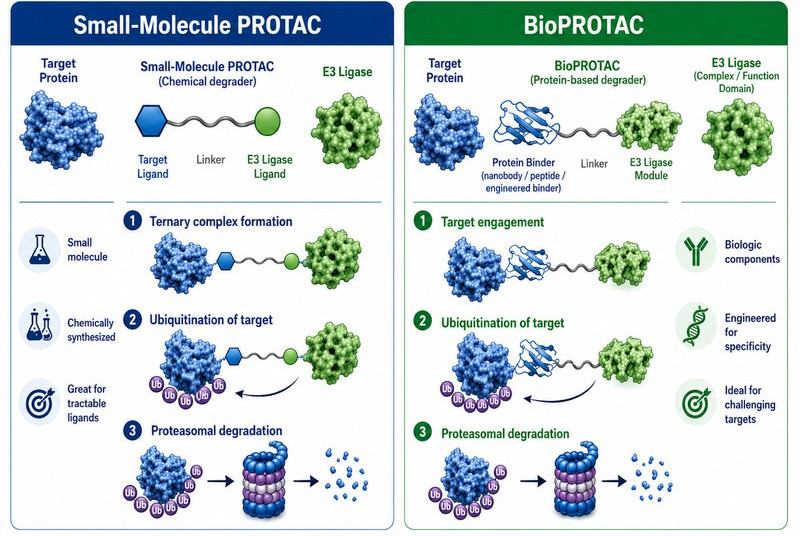 Fig.1 BioPROTAC and Small-Molecule PROTAC Comparison (BOC Sciences Original).
Fig.1 BioPROTAC and Small-Molecule PROTAC Comparison (BOC Sciences Original).
Shared Logic: Induced Proximity for Protein Degradation
Both approaches treat proximity as a functional signal. The degrader does not need to permanently occupy the target or block every active site. Instead, it must create a transient but productive arrangement that enables ubiquitin transfer. This event-driven logic can remove target proteins with catalytic, scaffolding, control, or structural roles, making targeted protein degradation useful for research questions that cannot be addressed by classical inhibition alone.
Structural Differences Between BioPROTAC and PROTAC
Although BioPROTAC and small-molecule PROTAC share the same induced-proximity concept, their molecular formats create different design priorities. A small-molecule PROTAC is built through chemical assembly of a target ligand, linker, and E3 ligase ligand, so optimization usually focuses on linker SAR, ternary complex cooperativity, cell permeability, metabolic stability, and physicochemical properties. In contrast, BioPROTAC is typically engineered from biological recognition modules, such as peptides, nanobodies, monobodies, or protein binders, fused to an E3 ligase-related component. This makes BioPROTAC especially useful when a target lacks a suitable small-molecule ligand, but it also shifts attention toward binder quality, fusion orientation, protein expression, intracellular delivery, and subcellular localization.
Table 2. Comparison of BioPROTAC and Small-Molecule PROTAC Strategies.
| Feature | BioPROTAC | Small-Molecule PROTAC |
|---|
| Core format | Engineered protein, peptide, nanobody, monobody, or fusion-based degrader. | Chemical degrader composed of target ligand, linker, and E3 ligase ligand. |
| Target-recognition strategy | Uses biological binders that can recognize broad protein surfaces or conformational epitopes. | Requires a suitable chemical ligand or warhead for the target protein. |
| E3 recruitment | Often uses an E3 domain, E3 adaptor, or engineered E3-related fusion element. | Uses a small-molecule E3 ligand to recruit an endogenous E3 ligase complex. |
| Optimization focus | Binder selectivity, fusion orientation, linker peptide design, expression level, localization, and delivery. | Binary affinity, ternary complex cooperativity, linker SAR, permeability, stability, and exposure. |
| Typical research value | Rapid assessment of target degradability and protein-level biology when chemical ligands are limited. | Development of chemically defined degrader series with scalable analog optimization. |
When BioPROTAC May Be Preferred?
BioPROTAC may be preferred when the main research barrier is target recognition rather than chemical optimization. For proteins without a clear small-molecule binding pocket, such as transcription factors, scaffold proteins, intrinsically disordered proteins, or protein-protein interaction targets, a protein-based binder may provide a more practical starting point than searching for a conventional warhead. BioPROTAC is also suitable when a validated nanobody, monobody, peptide binder, or engineered binding domain is already available, because the binder can be directly connected to an E3 ligase-related module to test whether the target protein is degradable. In early target validation, this approach helps researchers answer whether forced proximity to a degradation machinery can reduce the target protein level before investing in a full small-molecule PROTAC discovery campaign.
BioPROTAC can also be useful when researchers need high modularity, fast construct iteration, or genetically controlled degradation in defined cellular systems. By changing the target-binding domain, E3 module, linker, localization signal, or expression strategy, researchers can compare multiple degradation architectures in parallel. This makes BioPROTAC especially valuable for mechanism studies, proof-of-concept degradation experiments, functional genomics, and exploratory work on difficult targets where ligand availability is limited.
When Small-Molecule PROTAC May Be Preferred?
Small-molecule PROTAC may be preferred when the target already has a suitable ligand or inhibitor that can be converted into a target-binding warhead, and when the research goal is to build a chemically defined degrader series. In this situation, medicinal chemistry can be used to adjust linker length, attachment position, E3 ligand selection, polarity, permeability, metabolic stability, and degradation potency. Small-molecule PROTACs are also better suited for experiments that require direct compound treatment rather than genetic expression or protein delivery, because researchers can control concentration, exposure time, washout, and structure-activity relationship analysis in a familiar chemical workflow.
This format is often preferred when the project has moved beyond proof-of-concept degradability and requires systematic lead optimization. If robust target ligands, E3 ligase ligands, and cellular degradation assays are available, small-molecule PROTAC development can support analog screening, degradation kinetics profiling, selectivity studies, and in vitro or in vivo evaluation. In many research programs, BioPROTAC and small-molecule PROTAC are complementary rather than competing strategies: BioPROTAC can help validate whether a target can be degraded, while small-molecule PROTAC can then be optimized as a chemically tractable degrader format.
Major Types of BioPROTAC Designs
BioPROTACs are not a single molecular format. They can be designed using different binder classes, E3-related modules, and control strategies. The best format depends on target biology, available binders, desired localization, expression method, and assay objectives.
Peptide-based BioPROTACs use a short peptide sequence to bind the target protein. Peptides are attractive because they are relatively easy to design, synthesize, mutate, and fuse to other protein domains. They can also mimic natural interaction motifs, which makes them useful for targets controlled by short linear motifs or modular interaction domains. However, peptide binders may have weaker affinity or lower selectivity than folded protein binders, and they may require careful optimization to avoid degradation of unrelated proteins that recognize similar motifs.
Nanobody-Based BioPROTACs
Nanobody-based BioPROTACs are among the most promising formats because nanobodies are compact, folded, and often capable of recognizing specific protein surfaces with high affinity. They can bind endogenous proteins, tagged proteins, or conformation-specific epitopes, depending on how they are selected. A nanobody can be fused to an E3-related domain to generate a modular degrader, and changing the nanobody can redirect the degradation system to a new target. This makes nanobody-based BioPROTACs valuable for target validation, functional genomics, and the study of proteins that lack well-defined chemical ligand pockets.
Monobody and Designed Binder-Based BioPROTACs
Monobodies and computationally designed binders can recognize protein surfaces that are difficult for small molecules to engage. These binders can be selected or engineered for desired epitopes, and they can be fused to E3-related modules to test degradation. Because binder geometry influences ubiquitination efficiency, designed binders offer an opportunity to tune not only affinity but also the spatial relationship between the target and E3 module. In practice, multiple binders against different epitopes may need to be compared to identify the geometry that supports the deepest and most selective degradation.
AdPROM and Ligand-Inducible BioPROTAC Systems
Affinity-directed protein degradation systems use intracellular binders, such as nanobodies or monobodies, to direct E3-related activity toward a target protein. Ligand-inducible versions add another layer of control by allowing degradation to be switched on or adjusted through a separate input. These systems are useful for research programs that require temporal control, reversibility, or comparison between sustained and transient target depletion. They also demonstrate how BioPROTAC thinking extends beyond a fixed fusion protein and into programmable degradation circuits.
Switchable and Logic-Gated BioPROTAC Systems
Switchable BioPROTAC systems are an emerging direction in targeted protein degradation research. Instead of designing a degrader that is continuously active, researchers can engineer systems in which degradation depends on cleavage, ligation, localization, or the presence of a second molecular input. This approach is attractive for synthetic biology, pathway analysis, and dynamic protein network studies because it allows protein abundance to be modulated with timing and context. Future BioPROTAC platforms may therefore operate not only as degraders, but also as tunable protein-control systems.
Need a Custom BioPROTAC for Your Target Protein?
BOC Sciences provides customized BioPROTAC-related support, including target binder strategy, E3 ligase system design, fusion architecture planning, and degradation assay development for your research needs.
Get Consultation
Design Principles for BioPROTAC Development
BioPROTAC development requires integrated design across protein engineering, degradation biology, and assay development. Each module should be evaluated in isolation and in the final fusion context. A strong target binder, a functional E3 module, and a reasonable linker do not automatically produce degradation; they must form a productive spatial arrangement in the correct cellular compartment.
Selecting the Target-Binding Domain
The target-binding domain should be selected according to affinity, selectivity, epitope accessibility, intracellular folding, and compatibility with the intended fusion architecture. When several binders are available, it is useful to test binders against different epitopes. One epitope may place the target in a position that supports ubiquitin transfer, while another may orient the target away from the E3 module. For endogenous proteins, binders should be tested in the relevant cell background because native protein complexes, post-translational modifications, and localization can influence accessibility.
Choosing the E3 Ligase or E3 Functional Domain
E3 selection is central to BioPROTAC performance. The chosen E3-related module should be active, compatible with the target compartment, and capable of supporting ubiquitin transfer in the desired experimental system. BioPROTAC research can explore E3 domains beyond the small set commonly used in small-molecule PROTAC chemistry, but each new E3 design must be validated carefully. Researchers should confirm that target reduction depends on the E3 module, the target binder, and proteasomal degradation rather than indirect effects on transcription, translation, or protein stability.
Optimizing Fusion Orientation and Linker Design
Fusion orientation can dramatically change degradation efficiency. A target binder fused to the N-terminus of an E3 module may behave differently from the same binder fused to the C-terminus. Linker length, flexibility, rigidity, and amino acid composition can influence folding, steric accessibility, and proximity geometry. A linker that is too short may prevent productive contact; a linker that is too long may reduce effective proximity or increase conformational entropy. Practical BioPROTAC optimization often involves a small panel of linker lengths and orientations before deeper mechanistic assays are performed.
Engineering Degradation Efficiency and Selectivity
BioPROTAC efficiency can be improved by optimizing binder affinity, expression level, E3 domain activity, subcellular localization, and target accessibility. Selectivity should be assessed at both the target-family level and proteome level where possible. Because protein binders may recognize structured epitopes, selectivity can be excellent, but off-target interactions may still occur if similar surfaces exist in related proteins. Negative controls such as non-binding binder variants, inactive E3 modules, and unrelated target binders can help distinguish specific degradation from cellular stress or overexpression artifacts.
Recommended BOC Sciences Support for BioPROTAC Design
BOC Sciences provides integrated support for degrader design, binder strategy, E3 ligase selection, linker architecture, structural modeling, and assay planning. These services can help researchers build a rational BioPROTAC workflow from target selection to degradation validation.
Table 3. Recommended Services for BioPROTAC Design and Optimization.
| Service Name | Description | Inquiry |
|---|
| PROTAC Design Services | Supports integrated degrader design, including target selection, E3 recruitment strategy, linker architecture, and optimization planning for PROTAC and BioPROTAC-related research. | Inquiry |
| Ligand Design for Target Protein | Helps identify or optimize target-binding elements that can support target recognition in degrader design workflows. | Inquiry |
| Design of the Ligase System | Supports selection and design of E3 ligase recruitment strategies based on target biology, localization, and degradation mechanism. | Inquiry |
| Linker Design and Optimization Services | Optimizes linker length, composition, flexibility, rigidity, and attachment geometry for productive degrader architecture. | Inquiry |
| Protein Structure Modeling | Provides structural modeling support to evaluate binder-target interactions, domain orientation, and possible fusion architectures. | Inquiry |
BioPROTAC Delivery and Expression Strategies
Because BioPROTACs are protein-based or biologically encoded systems, delivery and expression are central design challenges. The degrader must reach the correct intracellular compartment, maintain its folded state, and be present at an appropriate level relative to the target. Different research goals may require different formats, including plasmid expression, viral expression systems, inducible expression, recombinant protein delivery, lipid-mediated delivery, or other cellular introduction methods.
Genetically Encoded BioPROTAC Expression Systems
Genetically encoded expression is one of the most direct ways to study BioPROTAC function. Researchers can introduce a construct encoding the target-binding module, linker, and E3-related module, then monitor target protein levels over time. Inducible expression systems are useful when continuous degrader expression may complicate interpretation. They allow comparison between baseline target abundance and degrader-induced target loss, while also enabling time-course analysis of degradation and recovery. However, expression level must be controlled carefully because overexpression can create non-physiological stoichiometry or overload protein quality-control pathways.
Recombinant BioPROTAC Protein Delivery
Recombinant BioPROTAC delivery aims to introduce the degrader protein directly into cells rather than relying on intracellular expression. This approach can improve temporal control because the amount and timing of degrader exposure can be adjusted experimentally. The major challenge is intracellular delivery: most proteins do not readily cross cellular membranes or escape endosomal compartments. Research strategies therefore focus on delivery vehicles, protein surface engineering, charge modification, and formulation approaches that improve cytosolic availability while preserving degrader activity.
Lipid-Mediated and Nanoparticle-Assisted Delivery
Lipid-mediated delivery has become an important direction for recombinant BioPROTAC research. Engineered BioPROTAC templates can be designed to interact with lipid systems, supporting intracellular delivery and rapid target degradation in cellular assays. This approach is useful for evaluating proteins located in the cytosol, nucleus, membrane-associated compartments, or other accessible regions, provided that the delivered degrader reaches the correct site of action. For research workflows, lipid-mediated delivery may also provide a useful comparison with genetically encoded expression because it separates degrader dose from transcriptional control.
Subcellular Localization Considerations
Subcellular localization can determine whether a BioPROTAC succeeds or fails. A nuclear target may require a nuclear localization sequence, while a cytosolic target may not. Membrane-associated targets may require orientation-sensitive binding and careful consideration of which protein domains are exposed to the cytosol. Mitochondrial or organelle-associated targets introduce additional constraints related to import, localization signals, and E3 availability. Early localization analysis by fluorescence imaging, fractionation, or proximity assays can prevent misinterpretation of a poorly localized BioPROTAC as an ineffective degradation design.
BioPROTAC Evaluation and Assay Development
BioPROTAC evaluation should answer five core questions: does the binder engage the target, does the E3-related module remain functional, does the fusion degrader reduce target protein level, is the reduction proteasome-dependent, and is the effect selective? A robust assay cascade should combine binding studies, cellular degradation assays, mechanistic validation, and selectivity analysis.
Target Engagement and Binder Validation
Target engagement can be evaluated using biochemical binding assays, co-immunoprecipitation, pull-down analysis, cellular colocalization, proximity labeling, or reporter systems. The exact method depends on the binder format and target biology. For BioPROTACs, binder validation should preferably be performed both before and after fusion to the E3-related module, because fusion may alter folding, affinity, or epitope accessibility. When possible, binding should be compared with a non-binding mutant or unrelated binder control.
BioPROTAC Expression and Stability Analysis
The BioPROTAC itself should be monitored. Researchers should confirm expression, molecular size, integrity, localization, and stability. If the degrader is unstable, degraded, mislocalized, or poorly expressed, target degradation may be weak even when the design concept is sound. Western blotting, fluorescence tagging, mass spectrometry, and protein stability assays can be used to evaluate the degrader construct. For recombinant delivery, assays should also assess cytosolic entry and persistence over the experimental time course.
Target Protein Degradation Assays
Target protein degradation is commonly measured by Western blotting, ELISA, immunofluorescence, flow cytometry, reporter degradation assays, or quantitative proteomics. In BioPROTAC workflows, time-course experiments are particularly useful because genetically encoded degraders may require time for expression before target loss becomes visible. Key readouts include degradation depth, degradation rate, residual target abundance, recovery after degrader removal or expression shutoff, and concentration- or expression-dependence. In in vitro assay systems, purified components can help dissect ubiquitination steps, while cellular systems reveal whether the complete degradation pathway operates in the biological context.
Mechanistic Validation of Ubiquitin-Proteasome Dependence
Mechanistic validation is essential. Target reduction should be rescued or altered when proteasome activity is blocked, when the E3 module is inactivated, or when the target-binding module is disrupted. Ubiquitination assays can provide direct evidence that the target receives ubiquitin chains after BioPROTAC expression or delivery. Additional controls may include measuring target mRNA, using unrelated proteins as selectivity controls, comparing different E3 modules, and testing whether degradation requires expected binding interfaces.
Selectivity and Off-Target Assessment
Selectivity should be evaluated beyond the target protein alone. Related protein family members, known binding partners, and broader proteomic changes can help determine whether the BioPROTAC acts as intended. Because BioPROTACs are often expressed as fusion proteins, researchers should also monitor cellular stress markers, protein quality-control pathways, and localization changes that might indirectly affect protein abundance. A selective BioPROTAC should show target reduction that is consistent with its binding specificity and degradation mechanism.
Recommended BOC Sciences Support for BioPROTAC Evaluation
BOC Sciences offers assay services that can support BioPROTAC research from target engagement to degradation validation and ubiquitination mechanism analysis.
Table 4. Recommended Services for BioPROTAC Evaluation.
Applications of BioPROTAC in Protein Degradation Research
BioPROTACs are useful research tools wherever direct protein-level depletion provides clearer insight than gene knockdown, overexpression, or classical inhibition. Their modularity allows researchers to probe target degradability, compare E3 systems, evaluate binder geometry, and study the consequences of rapid protein depletion.
BioPROTACs for Undruggable and Non-Enzymatic Proteins
Many proteins of interest do not have enzymatic active sites. Transcription factors, scaffolding proteins, adaptor proteins, aggregation-prone proteins, and proteins involved in large interaction networks may be difficult to inhibit with small molecules. BioPROTACs can approach these proteins from a different angle by using a biological binder against an exposed surface. If degradation is achieved, the entire protein can be removed, reducing both catalytic and non-catalytic functions. This makes BioPROTACs attractive for exploring proteins whose biological role depends on abundance, localization, or complex formation.
BioPROTACs for Functional Genomics and Target Validation
Protein-level depletion can produce different insights from RNA-level knockdown. RNA interference and gene editing act upstream of protein expression, while BioPROTACs act directly on the protein product. This difference is important when proteins are stable, when protein complexes buffer expression changes, or when rapid degradation is required to study acute functions. BioPROTACs can help determine whether target removal produces the expected cellular phenotype, whether partial degradation is sufficient, and whether a target remains biologically important in a specific context.
BioPROTACs for Synthetic Biology and Conditional Protein Control
Because BioPROTACs are protein-engineered systems, they can be integrated into synthetic biology workflows. Researchers can design systems that respond to expression inputs, localization signals, protease activity, split-domain reconstitution, or logic-gated control. Such designs can support experiments in which protein degradation must occur only under defined cellular states or time windows. This moves targeted degradation from a static intervention toward a programmable control layer for protein networks.
BioPROTACs as Discovery Tools for Small-Molecule Degrader Design
BioPROTACs can also inform chemical degrader discovery. If a BioPROTAC successfully degrades a target, researchers gain evidence that the target can be removed through the ubiquitin-proteasome system. The results can guide the search for small-molecule warheads, identify favorable target epitopes, support E3 selection hypotheses, and prioritize assay readouts for later small-molecule PROTAC development. In this way, BioPROTAC can serve as a biological feasibility platform before chemical optimization begins.
Current Challenges in BioPROTAC Research
Although BioPROTACs expand the targeted degradation toolbox, they also introduce challenges that differ from chemical PROTACs. The major issues include intracellular delivery, construct size, expression level, fusion protein stability, E3 compatibility, target accessibility, and assay interpretation.
Intracellular Delivery and Cellular Uptake
Protein-based degraders must enter the cell and reach the correct compartment. Genetically encoded systems solve the entry problem by producing the degrader inside cells, but they require transfection, transduction, or engineered expression workflows. Recombinant protein delivery provides external control over degrader exposure, but it must overcome membrane entry and endosomal escape barriers. Delivery remains one of the most important technical areas for BioPROTAC platform improvement.
Protein Size, Stability, and Expression Control
BioPROTACs can be larger and more structurally complex than small-molecule degraders. Large fusion proteins may fold inefficiently, aggregate, localize incorrectly, or be degraded themselves. Expression level can also influence results: too little BioPROTAC may produce incomplete degradation, while too much may cause non-specific effects or alter target stoichiometry. Careful construct design, expression tuning, and direct measurement of BioPROTAC abundance are therefore essential.
E3 Compatibility and Target Accessibility
A target binder and an E3-related module must be compatible in space and time. The target protein must present accessible ubiquitination sites, and the E3 module must be able to reach them. Some targets may resist degradation because the bound epitope positions the E3 too far from suitable residues, or because the target resides in a complex that blocks ubiquitin transfer. Testing multiple binders, linkers, orientations, and E3 modules can improve the chance of identifying a productive configuration.
Translating Proof-of-Concept into Robust Research Workflows
BioPROTAC proof-of-concept experiments must be converted into reproducible workflows. This requires well-defined controls, validated target antibodies or detection reagents, expression-normalized analysis, replicate assay formats, and selectivity evaluation. A well-designed BioPROTAC workflow should distinguish true targeted degradation from indirect changes in protein expression, cellular stress, or assay artifacts.
Future Directions of BioPROTAC Technology
BioPROTAC technology is moving toward more modular, programmable, and delivery-compatible systems. Future progress will likely come from advances in binder discovery, computational protein design, E3 ligase engineering, intracellular delivery, and dynamic control of degradation activity.
AI-Assisted Binder Design for BioPROTACs
Computational protein design can accelerate the discovery of target-binding domains that recognize selected epitopes or conformational states. AI-assisted modeling may help predict binder-target interfaces, fusion orientation, linker flexibility, and possible steric conflicts. These tools will not replace experimental validation, but they can narrow design space and help prioritize constructs for synthesis and testing.
Expanding the E3 Ligase Toolbox
BioPROTACs provide an opportunity to explore a broader set of E3 ligase-related modules than those currently supported by small-molecule recruiters. Expanding the E3 toolbox may improve degradation in specific compartments, cell types, or target classes. The most useful E3 systems will be those that combine robust activity, manageable expression, target-compatible localization, and predictable selectivity.
Modular, Switchable, and Cell-Context-Aware BioPROTAC Platforms
Future BioPROTAC platforms may be designed for conditional activity, reversible target control, or multi-input logic. Instead of continuously degrading one target, a system could be engineered to degrade a protein only after a defined input, to switch between targets, or to combine degradation with other protein-control modules. This direction is especially relevant for studying dynamic signaling pathways and complex protein networks.
References
- Békés, Miklós, David R. Langley, and Craig M. Crews. "PROTAC Targeted Protein Degraders: The Past Is Prologue." Nature Reviews Drug Discovery, 2022. https://www.nature.com/articles/s41573-021-00371-6
- Burslem, George M., et al. "BioPROTACs as Versatile Modulators of Intracellular Therapeutic Targets Including Proliferating Cell Nuclear Antigen." Proceedings of the National Academy of Sciences, 2020. https://www.pnas.org/doi/10.1073/pnas.1920251117
- Chan, Alvin, et al. "Lipid-Mediated Intracellular Delivery of Recombinant bioPROTACs for the Rapid Degradation of Undruggable Proteins." Nature Communications, 2024. https://www.nature.com/articles/s41467-024-50235-x
- Simpson, Lorna M., et al. "Inducible Degradation of Target Proteins through a Tractable Affinity-Directed Protein Missile System." Cell Chemical Biology, 2020. https://www.cell.com/cell-chemical-biology/fulltext/S2451-9456(20)30236-1
- Zhou, Runhua, et al. "Targeted Degradation of Endogenous YAP by Nanobody bioPROTAC Inhibits Tumor Progression." Nature Communications, 2025. https://www.nature.com/articles/s41467-025-64426-7
- "Synthetic Protein Degradation Circuits Using Programmable Cleavage and Ligation by Sortase A." Nature Communications, 2025. https://www.nature.com/articles/s41467-025-63819-y
Our Support
BioPROTAC and PROTAC Research Support at BOC Sciences
BOC Sciences provides integrated products and services for targeted protein degradation research, supporting BioPROTAC concept design, target binder strategy, E3 ligase system planning, linker optimization, ubiquitination analysis, and degradation assay development. Our team can help researchers connect protein engineering concepts with practical PROTAC and BioPROTAC evaluation workflows.

BioPROTAC Design and Target Binder Strategy
E3 Ligase System and Ubiquitination Support
Linker, Fusion Architecture, and Delivery Strategy
BioPROTAC Evaluation and Degradation Assays
Frequently Asked Questions (FAQ)

Still have questions?
Contact Us
What is BioPROTAC in protein degradation?
BioPROTAC, or biological PROTAC, is a targeted protein degradation strategy that uses engineered biological binders such as peptides, nanobodies, antibody fragments, or protein domains to recruit a protein of interest to an E3 ligase-related degradation machinery. Compared with small-molecule PROTACs, BioPROTACs can be especially useful when the target lacks a high-quality small-molecule ligand or belongs to a protein class traditionally considered difficult to drug. This makes BioPROTACs a valuable complementary platform for exploratory degradation biology and early target validation.
How does BioPROTAC differ from traditional PROTAC?
Traditional PROTACs are usually small heterobifunctional molecules composed of a target-binding ligand, an E3 ligase ligand, and a linker. BioPROTACs follow the same induced-proximity logic but replace one or more chemical ligand components with biological recognition elements, such as nanobodies, peptides, or engineered protein binders. This difference expands the target space, because biological binders can recognize flat, extended, or protein-protein interaction surfaces that may be challenging for small molecules. For drug discovery teams, BioPROTACs are often used alongside PROTACs to compare degradation feasibility, target selectivity, and mechanism-driven design options.
Which targets are suitable for BioPROTAC development?
BioPROTAC development is particularly attractive for intracellular proteins that are difficult to address with conventional small molecules, including scaffold proteins, transcription-related proteins, protein-protein interaction nodes, mutant proteins, and proteins with limited ligandability. A strong BioPROTAC program usually begins with a validated binder, a rationally selected E3 ligase component, and assays that confirm target engagement, ubiquitination, and degradation. BOC Sciences can support customers with peptide and protein binder design, recombinant protein preparation, target-binding assay development, degradation assay support, and custom research workflows for early BioPROTAC feasibility studies.
What are key challenges in BioPROTAC design?
Key BioPROTAC design challenges include intracellular delivery, binder affinity, binder orientation, E3 ligase selection, linker architecture, expression format, degradation efficiency, and target selectivity. Unlike conventional small-molecule PROTACs, BioPROTACs may require careful engineering of protein size, stability, localization signals, and delivery-compatible formats. Small structural changes can strongly affect ternary-complex formation and downstream degradation. BOC Sciences can help drug development teams evaluate these variables through custom peptide synthesis, protein engineering support, cell-based degradation assays, target engagement studies, and analytical characterization tailored to BioPROTAC discovery needs.
Why use BioPROTAC alongside PROTAC discovery?
BioPROTACs are not intended to replace small-molecule PROTACs; instead, they provide a complementary route when chemical ligand discovery is slow, target surfaces are difficult to bind, or rapid biological validation is needed. A BioPROTAC can help researchers determine whether degrading a target is biologically meaningful before committing to extensive small-molecule optimization. This parallel strategy can de-risk early discovery by separating target-degradation feasibility from small-molecule developability. For teams building targeted protein degradation pipelines, combining BioPROTAC and PROTAC approaches can create a broader, more flexible platform for target prioritization and mechanism exploration.
Explore More
Discover More Research Products
Explore featured products that can expand your research options and accelerate your next discovery.
Expert Services to Move Your Project Forward
Access end-to-end service solutions that help bring efficiency, flexibility, and expertise to your research pipeline.
News
Technical Information

