Nitrogen-rich linkers have become a modular approach to packing multiple design criteria (solubility, conformational restriction, targeted bioactivity, etc.) into one low-molecular-weight entity. By tethering basic or heteroaromatic nitrogens at set intervals, such spacers can be protonated in acidic endosomes, participate in water-bridged H-bonds at the protein interface, and avoid oxidative cleavage in vivo. Recent series show that even a single nitrogen can invert isoform selectivity or enable oral exposure without increasing linker length, indicating that nitrogen density is now another knob to tune alongside length/flexibility.
Introduction
A defining aspect of this new wave is the use of nitrogen-rich linkers, a break from the classic notion of a linker as a simple hydrocarbon strap. With the addition of saturated amines, spiro-piperidines or conjugated triazoles, nitrogen confers pH-dependent charge, lone-pair directionality and metabolic soft spots to tune each step in the degradome. In turn, medicinal chemists can tune solubility, improve ternary-complex stability or alter tissue distribution all with a single synthetic step, accelerating the path from biochemical hit to orally active degrader.
What Are Nitrogen-Rich Linkers?
Nitrogen-rich linkers are spacer segments where a high proportion of the heavy atoms are nitrogen, whether in the form of basic aliphatic amines, heteroaromatic rings or fused azacycles. Simple examples include piperazine or piperidine units that introduce a basic center whose pKa can be tuned by remote electron-withdrawing groups, while more sophisticated constructs combine multiple nitrogens—imidazole, triazole, pyrimidine—into a conjugated rod that is planar yet polar. A unifying structural element is an asymmetric basicity: one nitrogen is alkylated to quench charge, the other is free to be protonated, creating a pH-sensitive base, which experiences membrane potentials upon endosomal trafficking. The molecular backbones are synthetically accessible: most building blocks are commodity chemicals that participate in Suzuki, Buchwald–Hartwig or click couplings, all of which are amenable to aqueous conditions. This makes late-stage diversification possible, with little protecting-group gymnastics required. Metabolically, the electron-withdrawing nitrogen lowers the electron density of adjacent carbons, which makes the linker less susceptible to hydroxylation, and more prone to innocuous N-oxidation. This molecular structure functions as a single envelope that simultaneously encodes solubility properties along with rigidity and molecular recognition capabilities.
Why Nitrogen Plays a Key Role?
Nitrogen sits at a nexus in the chemical periodic table, being both small enough to fit into an aromatic ring without angle strain, and electronegative enough to upset π-electron distribution and accept hydrogen bonds. This sets up three pharmacological knobs for tuning. First, the issue of basicity. The nitrogen lone pair can be protonated in acidic organelles to help escape membranes (being trapped there is not desirable) but remain uncharged in the cytosol, thereby increasing the therapeutic window without an enduring charge. Second, the directional influence of the lone pair, where the nitrogen acts as a weak hydrogen bond acceptor that can couple to backbone amides or otherwise well-ordered water molecules, thus providing a means of extending residence time without increasing MW. Third, the steering of metabolism, where N-oxidation is often a lower energy process than benzylic hydroxylation, can lead to the formation of polar metabolites that are easily excreted and have little propensity to form reactive electrophiles. In addition, the ability of nitrogen to engage in sp³ or sp² hybridisation provides the designer with a mechanism to switch between saturated conformational flexibility and aromatic planar rigidity within the same linker, an ability which carbon-only scaffolds can not provide. It is no longer sufficient to consider nitrogen a solubilising afterthought, but as a multi-tasking atom that tunes polarity, orientation and metabolic fate at the same time.
Advantages of Nitrogen-Rich Linkers
Nitrogen-rich linkers provide a bundled enhancement suite: pH-tunable charge, directional hydrogen-bond contacts and metabolic robustness are all condensed into a single, low-mass heterocyclic rod. By spacing out basic or aromatic nitrogens, chemists can increase polar surface area without the entropic cost of PEG chains, while the same lone pairs can serve as transient electrostatic staples that position the ternary complex into a catalytically-competent pose. Across several recent series these collective features have often translated high-affinity binders into bona fide cellular degraders, validating that nitrogen density is now a primary optimization handle rather than a solubility afterthought.
Enhanced Solubility and Polarity
Nitrogens embedded in the linker backbone represent an internal, pH-dependent solubilizing feature that can impart some of the benefits of PEGylation (extension of aqueous volume) or flexible alkyl chains (entropic gain) without incurring their liabilities (artificially inflated molecular weight or conformational entropy penalty). The protonation of aliphatic amines or piperazine groups in acidic endosomes can support membrane translocation, yet the groups remain largely uncharged at physiological pH values. The result is an expanded therapeutic window without a permanent charge. Nitrogens in heteroaromatic rings (pyridine, pyrazine, triazole) create an asymmetric dipole that can repel the perfectly-packed faces of a crystal with a high density of poly-phenyl rods. This results in a lower lattice energy and an increase in kinetic solubility in aqueous buffered solutions. This addition of polarity does not lengthen the conjugation unit, and so the improvement in dissolution rate is not cancelled out by a loss of photosensitivity or oxidative stability. Finally, the basic sites can be leveraged as salt-forming groups. Crystallization of a hydrochloride or maleate salt can improve dissolution by several orders of magnitude without degradation under ambient humidity. Because this protonation is fast and reversible, the charged form can facilitate intracellular trafficking without permanently capturing the drug in a cationic state that would otherwise be suboptimal for permeability. Taken together, these properties turn the linker into a self-contained solubility engine whose ionization state can be tuned by small changes to the structure rather than added formulation excipients.
Added Rigidity Through Ring Structures
Nitrogen insertion into aromatic or saturated rings results in a mechanically constrained backbone that preorganises the PROTAC into a low entropy shape conducive to simultaneous binding. Pyridine's sp2 hybridisation geometrically constrains the proximal atoms into a planar geometry which precludes rotation along the linker axis and therefore reduces the conformational search space that the warhead and ligase ligand must sample. Even saturated heterocycles like piperazine prefer a sterically locked chair conformation which represents a middle ground between the floppy plasticity of alkyl chains and the brittle overconstraint of purely aromatic rods. This "forgiving rigidity" results in faster on-rates and slower off-rates by biasing the construct towards orientations that keep the catalytic lysine in the ubiquitin-loading radius for a longer time. Critically, the rigidifying influence is not associated with the π-stacking promiscuity typically found in extended poly-phenyl systems; the electron-withdrawing nitrogen lowers the polarisability of the ring and makes it less apt to participate in off-target aromatic sandwiches. The constrained geometry also improves photostability, as the delocalized π-system quenches UV energy through internal conversion rather than the Norrish cleavage pathways seen in flexible chains. Overall, nitrogen-rich rings act as conformational capacitors that store geometric information during synthesis and unleash it upon binding, front-loading the entropic cost.
Improved Bioactivity Profiles
The results from the principles of pH-tunable solubility, directionality of hydrogen bonding, and conformational pre-organization described above culminate in an enhanced bioactivity profile with higher potency and improved metabolic stability. Cellular work shows that many nitrogen-containing degraders are able to achieve a persistent knock-down at lower exposures than hydrocarbon analogs, an effect that has been attributed to an increased residence time in the ternary complex. The basic nitrogen can also be a metabolic vulnerability – N-oxidation is typically a lower energy process than benzylic hydroxylation – leading to polar, water-soluble metabolites that are quickly excreted and have less propensity to generate reactive electrophilic species. Furthermore, the enhanced solubility limits colloidal aggregation, a frequent culprit for false positive cytotoxicity that can obscure real pharmacology. Lone pairs excel in distinguishing between isoforms based on minimal differences such as a single surface histidine or backbone carbonyl through electronic selectivity which hydrocarbon mimics fail to achieve. Finally, the presence of nitrogen atoms also allows for the possibility of orthogonal late-stage diversification, via quaternization, N-oxidation or salt formation, to allow an investigator to tune distribution, limit brain penetration or improve renal clearance without the need to redesign the rest of the molecule. When these attributes combine they produce a wider therapeutic index and stable pharmacokinetics while minimizing off-target liabilities which positions nitrogen-rich linkers as essential components of future degraders.
Common Nitrogen-Containing Linkers
Nitrogen-containing tethers are now the prototypical tool kit for optimizing rigidity, polarity and synthetic tractability in a single PROTAC spacer. Piperidine and morpholine provide saturated, conformationally biased rings that can be protonated on demand. In contrast, urea and carbamate moieties introduce two-point hydrogen-bond arrays that lock the ternary complex without contributing aromatic bulk. As each motif has a unique pKa, vector angle and metabolic fate, investigators can substitute one nitrogen motif for another until the degradation curve steepens and the therapeutic window expands.
Piperidine and Morpholine
Piperidine furnishes a chair-locked, six-membered cycle that acts as a molecular hinge: rotation about the equatorial C–N bond is frozen, but the ring can tilt by a few degrees to register the minor helix twist that is often present between warhead exit vector and ligase entry site. The secondary nitrogen can be alkylated or acylated without breaking chair geometry, furnishing an asymmetric handle that vectorises the ligand trajectory while leaving the distal nitrogen free for protonation. Morpholine switches one methylene for an oxygen, inserting an ether oxygen that lowers basicity by about half a pKa unit and increases the fraction uncharged at neutral cytosol, an attribute that enhances membrane permeability with no loss of water solubility. Crystallographic snapshots show that the ether oxygen can receive a hydrogen bond from backbone NH groups that are often present at the rim of the ternary interface, in effect stapling the PROTAC to the protein surface for a few extra milliseconds. Both rings lack benzylic C–H bonds which forces microsomal oxidation to target only the aliphatic chain near the carbonyl or nitrogen, creating polar N-oxides instead of reactive quinone-imines. Synthetic accessibility is a non-issue: Boc-protected piperidine and morpholine are stable shelf-sticks and the protecting group can be removed under mild acidic conditions that will not epimerise neighbouring stereocentres. Together, these attributes turn the saturated heterocycle into a conformationally biased, pH-responsive spacer that encodes rigidity and solubility without the π-stacking promiscuity that often comes with extended aromatic rods.
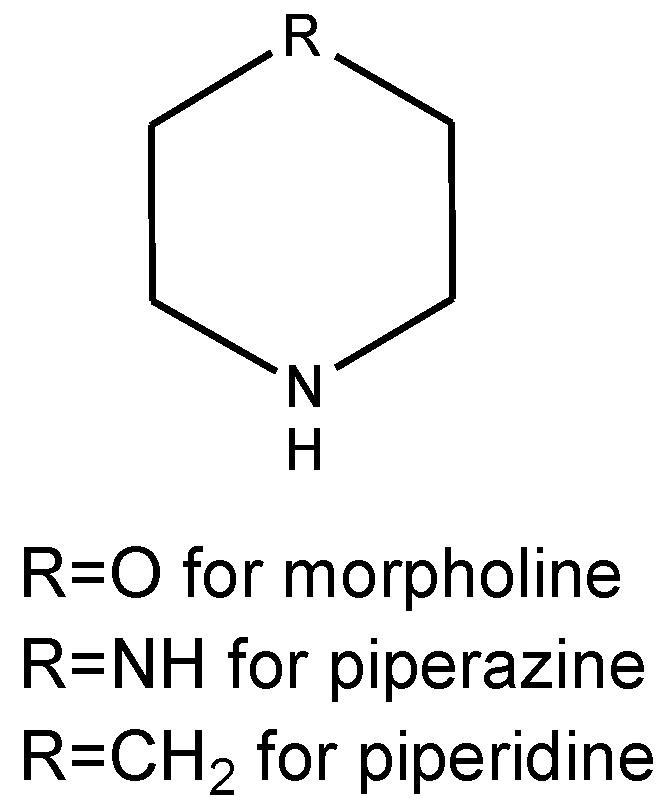 Fig. 1 Structure of morpholine, piperazine, and piperidine moieties.1,2
Fig. 1 Structure of morpholine, piperazine, and piperidine moieties.1,2
Urea and Carbamate-Based Linkers
Urea and carbamate motifs bring a two-point H-bond motif to bear, which acts as a molecular zipper across the target–ligase interface. The urea NHs are potential backbone carbonyl bridges, while the central carbonyl is a hydrogen bond acceptor for a nearby histidine or tyrosine, respectively. This creates a transient but directional staple that prolongs residence time without a commensurate increase in molecular weight. Carbamate is mono-donor, mono-acceptor by virtue of one NH being replaced with an oxygen atom, so it is less conformationally demanding but can also engage in water-bridged contacts when the interface is fringed with ordered solvent. Both motifs are intrinsically planar and so they impart rigidity along the linker axis, while remaining slender enough to fit within the tight grooves that are often found between the POI and the E3 ligase. From a metabolic perspective, the electron-deficient carbonyl is disfavored by oxidative metabolism; instead the linker is hydrolyzed by enzymes to give an amine and a CO2-containing fragment, routes which lead to innocuous metabolites that can be readily excreted. Synthetic installation is orthogonal: the isocyanate necessary for urea formation can be generated in situ from a nitro precursor, while the carbamate is accessible by alcohol-mediated coupling to an activated carbonate, reactions that are generally functional group tolerant and that can be performed on gram scale without chromatography. Taken together, urea and carbamate tethers serve as H-bond-encoded spacers that lock the ternary complex into a catalytically competent pose, while also providing a soft metabolic exit strategy.
Applications in PROTAC Research
Nitrogen-rich linkers have transitioned from academic curiosities to validated enablers of cellular degradation, with documented successes spanning kinases, epigenetic readers and DNA-repair enzymes. By swapping flexible alkyl chains for piperazine, triazole or pyridyl segments, researchers have repeatedly rescued oral exposure, shortened linker length and steepened degradation curves without redesigning warhead or ligase ligand. Conversely, emerging reports also catalogue bell-shaped dose responses, unforeseen off-target stacking and solubility ceilings that accompany excessive nitrogen density, underscoring that the same lone pair that rescues one programme can derail another if regiochemistry or basicity is mispaired.
Case Studies and Literature Examples
In an unrelated anti-cancer campaign against PARP1, an alkyl tether was exchanged for a rigid pyridine–piperazine hybrid that both reduced the end-to-end tether length and added a basic nitrogen adjacent to the ligase-facing amide. Crystallography confirmed the piperazine nitrogen to be engaged in a hydrogen bond with a backbone carbonyl of the E3 ligase, a contact that is missing in the flexible parent, and wash-out assays in cells showed prolonged degradation even after 24 h of nanomolar dosing. In another project, a triazole-rich linker was constructed via copper-free click chemistry to enable degradation of an oncogenic kinase. Triazole nitrogens engaged in a water-bridged network with a surface arginine on the target to position the catalytic lysine into the ubiquitin-loaded E2 active site; the resulting degrader was also active against a gate-keeper mutant that had confounded previous analogues. A third example used a morpholine–urea combination to degrade a DNA repair helicase. The urea NH donors bridged two backbone carbonyls at the target–ligase interface, and the morpholine oxygen provided a solubility-boost sufficient to enable oral dosing in a rodent model. The common theme in these otherwise disparate systems is that nitrogen was not merely included for solubility purposes but was placed to make new, weak but directional contacts that held the ternary complex together long enough for poly-ubiquitin transfer.
Potential Limitations
Nitrogen-rich linkers are not without potential liabilities that can negate these benefits if not carefully optimized. Too much basicity can cause lysosomal trapping, where the protonated species accumulates in acidic vesicles, contributing to the intracellular total load but not to the cytosolic free concentration. Regiochemical misplacement of the heteroatom can seed off-target π-stacking with ATP-binding sites or heme centres, generating apparent inhibition that is falsely interpreted as degradation. A second issue is the "hook effect": at high concentrations, the PROTAC saturates either the target or the ligase individually, precluding formation of the productive ternary complex and resulting in bell-shaped dose–response curves that make it difficult to identify an appropriate dose. Nitrogen-rich linkers can exacerbate this problem by increasing the binary affinity and shifting the bell maximum to lower concentrations, thereby unwittingly narrowing the therapeutic window. Metabolically, iterative N-oxidation or dealkylation can produce zwitterionic metabolites that are cleared quickly but still possess enough affinity to contribute to off-target binding, as was observed when piperazine linkers were coupled to electron-rich warheads. Finally, crystal packing energy often increases when multiple heteroatons are introduced, resulting in hygroscopic solids that require salt screening or amorphous dispersion to ensure consistent oral exposure. These pitfalls highlight that nitrogen incorporation must be counterbalanced by attention to basicity, regiochemistry and total lipophilicity; otherwise the same lone pair that salvages solubility can catalyse its own downfall.
Supply and Customization
At BOC Sciences, we supply a broad range of nitrogen-rich linkers designed to optimize solubility, rigidity, and overall bioactivity in PROTAC and small-molecule drug discovery programs. Our linkers are produced under strict quality standards and supported by analytical documentation, ensuring reproducibility and performance in every synthesis.
High-Purity Variants Ready to Order
We maintain a comprehensive inventory of ready-to-ship nitrogen-rich linkers, including:
- Piperidine, morpholine, pyrrolidine, urea, carbamate, and amidino-based scaffolds.
- High purity, verified by HPLC, LC-MS, and NMR to ensure structural accuracy and consistency.
- Pre-functionalized variants featuring reactive handles such as amine, azide, alkyne, and NHS for rapid conjugation.
- COA and analytical data packages included with every batch for traceability and compliance.
Our global warehouses ensure fast, temperature-controlled delivery, with standard shipping times for stocked items. Each product is carefully packaged to maintain integrity and stability during transit.
Custom Nitrogen-Rich Linker Design
For researchers developing specialized PROTACs or advanced degraders, we offer custom nitrogen-rich linker design and synthesis services. Our chemists collaborate closely with your R&D team to:
- Adjust ring size, flexibility, or polarity to achieve the right balance between permeability and solubility.
- Incorporate cyclic amines, bis-nitrogen systems, or tertiary amines for precise conformational control.
- Modify functional groups or spacer geometry to improve target-ligase orientation and cooperativity.
- Deliver isotopically labeled or GMP-adjacent batches for advanced pharmacokinetic or regulatory studies.
We provide rapid feasibility assessments, cost-effective synthesis routes, and full analytical verification (HPLC, LC-MS, NMR) with every order. Whether you need a single prototype or multi-gram scale production, our team ensures the highest standards of quality and reproducibility.
Partner with Us for High-Purity Nitrogen-Rich Linkers
Nitrogen-rich linkers play a vital role in tuning solubility, hydrogen bonding, and rigidity, making them indispensable in next-generation PROTAC and drug design. Selecting analytically verified linkers ensures consistent biological performance and predictable SAR outcomes. At BOC Sciences, we combine advanced synthesis capabilities, rigorous quality control, and global distribution to deliver nitrogen-rich linkers that meet your research and development needs. From standard piperidine derivatives to complex multi-nitrogen architectures, our chemistry delivers precision and reliability every time.
Contact our technical team today to request a quote, explore our stocked linkers, or initiate a custom nitrogen-rich linker synthesis for your next PROTAC program. Advance your discovery pipeline with linkers designed for balance, stability, and superior performance.
FAQs
1. Why use nitrogen-rich linkers in PROTACs?
They enhance solubility, introduce hydrogen-bonding capacity, and stabilize molecular conformation.
2. What nitrogen-rich structures are available?
Piperidine, morpholine, and urea-based linkers are among the most popular.
3. Can I design custom nitrogen linkers?
Yes, we offer tailored synthesis to adjust ring size, basicity, or polarity.
References
- Image retrieved from Figure 1 " Structure of morpholine, piperazine, and piperidine moieties," Zolotareva D.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Zolotareva D, Zazybin A, Dauletbakov A, et al. Morpholine, piperazine, and piperidine derivatives as antidiabetic agents[J]. Molecules, 2024, 29(13): 3043. https://doi.org/10.3390/molecules29133043.
- Kerru N, Gummidi L, Maddila S, et al. A review on recent advances in nitrogen-containing molecules and their biological applications[J]. Molecules, 2020, 25(8): 1909. https://doi.org/10.3390/molecules25081909.
- Kubryń N, Fijałkowski Ł, Nowaczyk J, et al. PROTAC Technology as a New Tool for Modern Pharmacotherapy[J]. Molecules, 2025, 30(10): 2123. https://doi.org/10.3390/molecules30102123.
