Heteroaryl linkers have emerged as more than mere tethers, acting as tools of directional control to tune polarity, dipole moment and inter-protein contacts within the ternary complex. Introduction of lone pairs, hydrogen-bond acceptors or weak donors by the simple expedient of swapping carbons for nitrogens, oxygens or sulfurs allows the linker to dock against backbone amides or structured water molecules, frequently re-orienting the whole chimera by a few degrees – enough to reposition a lysine side chain inside the catalytic cleft. Recent structure–activity surveys have shown that a single heteroatom swap can invert isoform selectivity or rescue degradation of targets that resisted purely hydrocarbon rods, making it clear that heteroaromatic rings are first-choice rather than fallback options.
Introduction
In contrast to benzenoid spacers, heteroaryl rings display electronically inhomogeneous surfaces that can be tuned atom-by-atom. The unshared electron pairs on pyridine nitrogen, for instance, decrease the quadrupole moment with respect to benzene, diminishing face-to-face π-stacking, but also provide an anchor for transient protonation or hydrogen bonding. With the ring remaining conjugated, the entropic advantage of pre-organization is retained, but the physicochemical signature – polar surface, dipole vector, pKa – can be tuned independently of length. Synthetic access is also highly versatile: cross-coupling, C–H activation or click chemistry can be used for the late-stage installation of thiazole, oxadiazole or pyrimidine motifs under mild, scalable conditions.
The Role of Heteroaryl Linkers in PROTACs
The linker is the only component inside the cell that interfaces with the bulk solvent and both the ligase and target surfaces simultaneously; hence embedding a heteroaryl unit creates multiple optimization opportunities. A pyridyl nitrogen can accept a hydrogen bond from a backbone NH that is too distant for direct contact, creating a water-bridged staple that lengthens residence time without increasing molecular weight. Furan or thiophene inserts a polarisable oxygen or sulfur that can engage in dispersive interactions with methionine or cysteine side chains, often filling a lipophilic cavity that is inaccessible to the flat face of benzene. The ring gains a molecular dipole due to the heteroatom interference with π-electron distribution which aligns with local electrostatic fields and subtly directs the warhead's exit vector to make the catalytic lysine face the E2-loaded ubiquitin. Regiochemistry provides an additional dial: moving the nitrogen from meta to para reorients the lone pair by sixty degrees, a geometric tweak that can switch a flat SAR curve into a steep sigmoid without touching the terminal ligands. Beyond binding, the heteroaryl segment modulates whole-molecule properties. The basicity of pyridine or imidazole can be tuned to become protonated inside acidic endosomes, promoting membrane release while remaining uncharged in the cytosol, thus improving oral exposure without permanent charge. Metabolically, the electron-deficient ring resists hydroxylation better than electron-rich phenyl, yet can be oxidized to N-oxide, a metabolite that is often more polar and more readily excreted than the corresponding phenol. Finally, the synthetic orthogonality of Suzuki, Sonogashira or click couplings allows heteroaryl halides or boronic acids to be swapped overnight, enabling rapid iteration that is difficult to achieve with heavily substituted alkyl chains. The combination of these properties enables heteroaryl linkers to function as multifunctional modules which show binding cooperativity while maintaining metabolic strength and physiochemical stability all within a single light-weight entity.
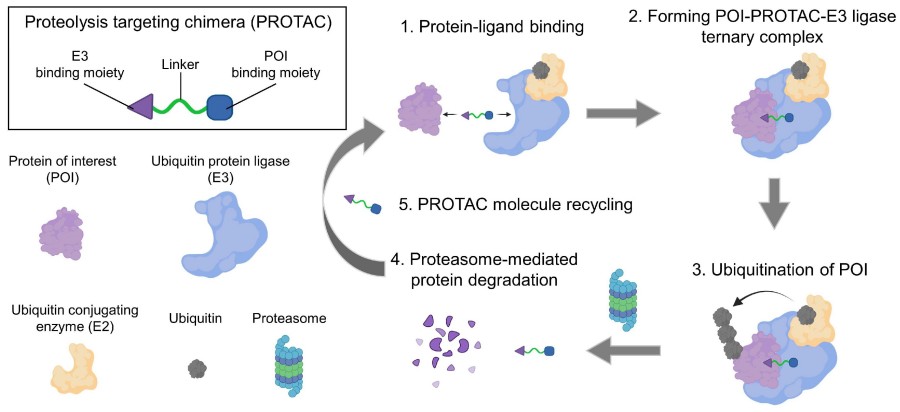 Fig. 1 Schematic illustration of the mechanism of PROTAC.1,2
Fig. 1 Schematic illustration of the mechanism of PROTAC.1,2
Differences from Aromatic Hydrocarbons
Compared to linker backbones that bear only hydrogen, aromatic hydrocarbons like phenyl or biphenyl impart rigidity and π-stacking, but lack the directional handles to tune ternary complex geometry. Phenyl imparts a symmetrical quadrupole moment that intercalates between protein aryl side chains without strong orientational preference. While this attribute may be a desired aspect in other circumstances (i.e. in narrow hydrophobic clefts) it provides no electronic compensation when the binding interface is flanked by charged or hydrogen bonding residues. Heteroaryl rings break this symmetry: the pyridine lone pair, the polarisable sulfur of thiophene or the weakly basic nitrogen of imidazole imbues a local dipole that can interact with backbone carbonyls or with ordered water molecules, 'stapling' the complex in a specific rotational register. Additionally, the heteroatom often reduces melting point and increases kinetic solubility versus an isosteric phenyl, mitigating the aggregation that often accompanies long, poly-aryl linkers. Photostability is another differentiating factor; phenyl rings can participate in photo-induced electron transfer with adjacent tryptophan residues, sometimes resulting in covalent adducts, while pyridine or triazole quenches UV energy via internal conversion, thereby minimising the incidence of photolytic side reactions. Metabolically, hydrocarbon linkers are vulnerable to oxidative hydroxylation that can produce reactive quinone-imines, whereas heteroaryl rings are more prone to N-oxidation or ring-opening pathways that often produce more polar, excretable metabolites. Synthetic flexibility is also different: A phenyl ring needs harsh electrophilic aromatic substitution conditions for fluorine or methoxy group introduction, but pyridine achieves similar electronic tuning through nucleophilic displacement or cross-coupling reactions that occur under mild conditions and maintain functional group integrity. Taken together, heteroaryl linkers do not simply mimic the rigidity of hydrocarbon analogues, they embellish it with directional bonding, pH-sensitive basicity and metabolic soft-spots that, collectively, open a larger optimization landscape for next-gen degraders.
Benefits of Heteroaryl Linkers
Heteroaryl linkers offer an opportunity to add multiple parameters to a rod without the increase in volume of a purely hydrocarbon chain, because they can encode directionality, tunable basicity and metabolic stability within the same aromatic area. The pyridine nitrogen, the polarizable sulfur of thiophene or the weakly basic nitrogen of imidazole serve as internal compasses: they steer the PROTAC towards local electrostatic fields, improve residence time through water-mediated interactions and increase kinetic solubility without the entropic cost of PEG chains. In series reported in recent years, these combined effects have repeatedly turned high-affinity binders into actual cellular degraders, and this is why heteroaryl moieties are now considered mandatory.
Polarity Control in Ternary Complexes
Inserting a heteroatom into the π-framework polarizes the molecule, creating a molecular dipole that can be directed by regiochemistry and provides an electronic fine-tune knob absent in hydrocarbons. For example, the pyridyl nitrogen withdraws electron density from the para carbon, concentrating lone-pair density at the heteroatom; this physical asymmetry creates a quadrupole moment that complements backbone carbonyls or with oriented water molecules, which are common at the margins of target–ligase interfaces. Dipole-aligned contacts act as a transient electrostatic velcro to hold the pieces of the nascent ternary complex together for a few extra milliseconds, long enough for the E2–ubiquitin thioester to come in range of the substrate lysine. Because the interaction is directional, it can compensate for minor angular mismatches in exit-vector without redesigning the ligand core. Regiochemistry is another lever, as sliding the nitrogen from meta to para rotates the negative end of the dipole by sixty degrees, a geometric tweak that can convert a flat SAR curve into a steep sigmoid without changing the linker length. The added polarity also softens the melting point of the solid PROTAC and improves kinetic solubility in buffered media, both consequences of a lower tendency toward colloidal aggregation seen with purely carbocyclic linkers. Partial protonation of the heteroatom at lysosomal pH also offers a cationic handle to support membrane escape, yet remain mostly shielded at neutral cytosolic pH to support oral exposure without permanent charge. These features in aggregate convert the linker from a passive spacer to an electronic actuator to tune binding cooperativity and physiochemical behavior.
Improved Orientation and Geometry
Heteroaryl rings provide an intrinsic vector: the lone pair or the heteroatom itself establishes a privileged direction within the π-cloud. Crystallographic snapshots of pyridine- or thiazole-containing PROTACs recurrently capture the nitrogen atom in position to point toward a backbone NH or a well-structured water molecule, behaving like a molecular compass that fixes the rotational register of the whole chimera. This "compass needle" effect is naturally missing in symmetrical phenyl rings, whose quadrupole moment is centrosymmetric and therefore provides no orientational preference. The geometric outcome is a reduced ensemble of bound poses: molecular dynamics trajectories show that heteroaryl-linked degraders sample a smaller number of rotational clusters and dwell longer in the productive basin, leading to faster ubiquitin transfer and more complete degradation at lower exposure. Regiochemistry tunes the compass: 3-pyridyl keeps the lone pair relatively shielded, reducing its inductive withdrawal from the conjugated pathway, whereas 4-pyridyl orients the dipole moment along the linker axis, a helpful adjustment when the ternary interface calls for a small electrostatic zipper. Thiophene and furan offer alternative vectors via their polarizable heteroatoms, allowing sulfur–π or oxygen–water contacts that augment the π-stacking interactions provided by the carbocyclic portion of the ring. The kinetic benefit is a slower off-rate that withstands transient dips in intracellular concentration, giving the ubiquitin machinery time to poly-merize the substrate even under wash-out conditions. In brief, the heteroatom converts a rotationally ambiguous rod into a geometrically programmed spacer that pre-orients the POI lysine toward the catalytic center.
Solubility Advantages
The same heteroatom that enforces orientation also counters the tendency of poly-phenyl rods to pack in crystal lattices of high symmetry, which depresses lattice energy and increases kinetic solubility. Pyridine, pyrazole or thiazole provide an asymmetric dipole that penalizes π–π stacking in the solid state. This effect can be strong enough to turn a crystalline, poorly dispersible powder into an amorphous, rapidly dissolving solid without the use of additional formulation excipients. The effect is enhanced when the heteroatom can be protonated; one basic nitrogen is sufficient to increase polar surface area enough to yield an improved dissolution rate, yet the lone moiety is low in mass to avoid the inflation in molecular weight that is a liability with PEGylation. Because the protonation event is fast and reversible, the charged species can help with escape from acidic endosomes without trapping the molecule in a cationic state that would adversely affect permeability. In addition, heteroaryl rings are narrower than their saturated heterocyclic analogues, so they take up less space in the often narrow cleft that is formed at the ternary interface, leaving more space for adaptive side-chain rearrangements. Heteroatoms that are electron-withdrawing lower the electron density of the neighbouring carbon framework, which in turn make the linker less prone to oxidative attack by cytochrome P450 enzymes. The slower clearance that results confers the dividend of steadier exposure. Taken together, these solubility and stability dividends enable heteroaryl linkers to provide the kinetic benefits of rigidity without the physicochemical liabilities that have long been associated with extended aromatic scaffolds.
Common Heteroaryl Types
Heteroaryl rings are electronically tunable, conformationally preorganized scaffolds that can be readily exchanged in PROTAC tethers without having to retrace back synthetic trees. Heteroaromatics range from six-membered π-deficient azines (pyridine, pyrimidine) to five-membered π-excessive azoles (imidazole, oxazole, thiazole), each of which comes with their own dipole moment, hydrogen-bond fingerprint and metabolic stability. Since the heteroatom is incorporated directly into the aromatic core, electronic effects are transmitted throughout the linker and enable chemists to tune basicity, π-stacking tendency and even pH-responsive switching within the same ring system. The subsequent subsections will illustrate how these nuanced electronics culminate into tangible differences in ternary-complex stability, cellular uptake and metabolic fate.
Pyridine and Pyrimidine
Pyridine furnishes one sp²-hybridised nitrogen which decreases the overall quadrupole moment of the ring (relative to benzene), making it less favorable for face-to-face π-stacking, while also furnishing a lone pair for hydrogen-bond acceptance. When the nitrogen is meta to the warhead, it can accept a backbone NH from the E3 ligase, providing additional enthalpy with no penalty to molecular weight; when it is para to the warhead, the resulting molecular dipole registers the degrader in an electrostatic groove, and thus "pre-shapes" the encounter complex. Pyrimidine (which has two nitrogens at the 1- and 3-positions) is more electron-deficient, and less basic (pKa ≈ 1 versus 5 for pyridine), and thus essentially neutral at physiological pH; this should avert off-target protonation in acidic tumour microenvironments. The second nitrogen also increases the number of canonical resonance forms, further stabilizing the ring toward oxidative metabolism, and decreasing the propensity for reactive epoxide formation. From a synthetic perspective, both rings are commercially available, with orthogonal handles (boronic acids, halogens, azides) that allow for convergent assembly under mild Suzuki or Sonogashira conditions. Taken together, pyridine and pyrimidine give an electronic gradient that ranges from mildly π-basic to distinctly π-acidic, giving medicinal chemists the ability to tune the π-stacking signature of the linker without changing its length or flexibility.
Imidazole, Oxazole, and Thiazole
Five-membered azoles are associated with a more acute dipole and smaller steric volume than six-membered azines and are thus well-suited for binding into a tight pocket where a larger ring system would be sterically hindered. Imidazole features two nitrogens in a 1,3-relationship, resulting in an amphoteric surface capable of acting as a hydrogen bond donor or acceptor depending on the tautomer and local pH; such switchable linkers are of value when a linker must straddle two proteins whose interface contains both donor and acceptor residues. Oxazole substitutes one nitrogen for an ether oxygen, resulting in a more electron-poor ring that is less susceptible to metabolic oxidation and provides a weaker hydrogen-bond acceptor when too much polarity would hinder membrane permeability. Thiazole makes the inverse substitution, replacing the oxygen with sulfur; this increases the polarizability and lengthens the π-system by virtue of sulfur d-orbital overlap, resulting in a ring that is more π-acidic and capable of stronger cation-π interactions with arginine or lysine side chains. Crucially, all three azoles are π-excessive compared to pyridine and are thus able to participate in offset π-stacking rather than face-to-face sandwiches, decreasing the likelihood of non-specific aggregation while still allowing for a measurable enthalpic benefit. Synthesis is direct: cyclodehydration of α-acylamino ketones (oxazole), condensation of α-halo aldehydes with thioamides (thiazole), or multicomponent reactions (imidazole) are all able to produce gram quantities in a single step, facilitating the rapid construction of azole-centric linker libraries for parallel screening.
Applications in Research
Heteroaryl linkers have evolved from structural curiosities to enabling elements throughout the entire targeted-degradation workflow. The electronically heterogeneous surfaces these linkers afford allow teams to fine-tune the permeability–aqueous solubility tradeoff while the built-in hydrogen-bond gates and π-clouds provide fresh points of contact within the ternary complex. The sections below review specific case stories and recurring traps that have come to define day-to-day optimization efforts.
Case Studies from Targeted Degradation
A canonical example describes how substitution of a flexible ether tether with a methylene–piperazine heteroaryl hybrid in a pirin-targeting PROTAC halved total polar surface area, sped cellular uptake and reduced the time to near-complete substrate depletion from 24 h to 2 h. The improvement was ascribed to a transient π-contact formed between the pyrimidine ring and a tyrosine residue on the ligase surface, an interaction not possible in the fully aliphatic analogue. In a related programme, introduction of a pyridyl–triazole linker between a bromodomain ligand and VHL-recruiting element was shown by cryo-EM to accept a backbone NH hydrogen bond from the ligase, buttressing an otherwise labile ternary assembly and reducing the intracellular concentration needed for detectable degradation. Use of a thiazole-centred linker against a hormone receptor demonstrated that the polarisability imparted by sulfur can fortify cation-π interactions with an arginine shelf, increasing selectivity over paralogue receptors even without additional warhead optimisation. Together, these examples show that heteroaryl tethers can rescue lost cellular activity from floppy or overly hydrophilic backbones while also refining the discrimination profile.
Challenges and Considerations
For all their virtues, heteroaryl linkers are not without liabilities that must be monitored. The most frequent is metabolic liabilities: oxidation can take place at multiple ring positions, which can lead to the formation of equipotent hydroxy metabolites that are difficult to account for when estimating clearance. Protonation of basic nitrogens is possible in acidic tumor microenvironments, which may enhance polarity and active efflux. The potential for light-activated radical formation also exists with electron-deficient rings, and such reactivity can lead to the color changes or adduct formation that can occur upon storage of these compounds. Cross-coupling reactions that are typically used to construct long heteroaryl backbones also can leave trace palladium or copper that require additional steps in purification. In addition, the same dipole moments that can enhance binding can also cause enhanced non-specific adsorption to glass or plastic surfaces, which is often observed when compounds are diluted in high throughput series. Thoughtful teams will counter these liabilities early in the discovery process by embedding heteroatom positions within a reliable SAR framework, validating metabolic maps in animal models, and confirming photosensitivity under typical laboratory conditions. When treated as an active pharmacophore instead of a passive spacer, the heteroaryl moiety can be used to exploit a unique electronic environment while avoiding the concerns that led to these motifs being relegated to rescue strategies in past efforts.
Ordering Information
At BOC Sciences, we provide a diverse selection of heteroaryl linkers designed to help researchers fine-tune polarity, orientation, and molecular geometry in targeted protein degradation (TPD) systems. Our linkers are analytically verified, readily available, and supported by custom synthesis services to meet the unique requirements of your PROTAC design.
Stock Availability and Functionalization
We maintain a robust catalog of ready-to-ship heteroaryl linkers, including:
- Pyridine, pyrimidine, imidazole, oxazole, and thiazole scaffolds.
- Pre-functionalized variants with reactive handles such as bromo, iodo, boronic acid (Bpin), azide, and alkyne.
- High-purity materials, validated by HPLC, LC-MS, and NMR testing.
- Comprehensive Certificate of Analysis (COA) and batch traceability for every product.
Custom Synthesis for Research Teams
If your PROTAC program demands a specific heteroaryl structure or unique substitution pattern, our custom synthesis service provides full flexibility. We can modify linker properties to match your project's design and pharmacokinetic goals, including:
- Custom ring substitutions (2-, 3-, or 4-position heteroatom placements).
- Hybrid aryl-heteroaryl frameworks for tuned dipole orientation and binding geometry.
- Polarity or lipophilicity adjustments via electronic modification or ring fusion.
- Custom end groups such as amine, azide, alkyne, NHS, or carboxyl to enable diverse conjugation strategies.
Our expert chemists collaborate directly with your R&D team to ensure optimal E3 ligase compatibility, ternary complex stability, and synthetic scalability. We also provide transparent lead times, detailed analytical reports, and global delivery with full documentation.
Partner with Us for High-Quality Heteroaryl Linkers
Heteroaryl linkers are essential tools for controlling polarity, electronic effects, and 3D orientation in PROTAC and TPD design. Selecting a well-characterized, high-purity linker can dramatically improve ternary complex formation, potency, and selectivity - making the difference between a lead and a breakthrough. At BOC Sciences, we supply analytically validated heteroaryl linkers and custom synthesis solutions to support every stage of your discovery pipeline. Whether you need pyridyl, oxazole, or thiazole linkers, we deliver precision chemistry with rapid turnaround and global reach.
Contact our technical specialists today to request a quote, explore our catalog, or discuss a custom heteroaryl linker synthesis tailored to your research goals. Empower your next-generation PROTAC program with linkers designed for performance, stability, and innovation.
FAQs
1. What are heteroaryl linkers used for?
They modulate polarity, orientation, and hydrogen-bonding within PROTAC ternary complexes.
2. Which heteroaryl scaffolds do you supply?
Pyridine, pyrimidine, imidazole, oxazole, and thiazole linkers are available off the shelf.
3. Can I order linkers with specific substitution patterns?
Yes, 2-, 3-, and 4-substituted heteroaryl variants can be customized to your design.
4. Do you offer analytical verification?
Each batch includes HPLC, LC-MS, NMR, and a COA.
References
- Image retrieved from Figure 1 " Schematic illustration of the mechanism of PROTAC," Liu Z.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Liu Z, Zhang Y, Xiang Y, et al. Small-molecule PROTACs for cancer immunotherapy[J]. Molecules, 2022, 27(17): 5439. https://doi.org/10.3390/molecules27175439.
- Catarzi D, Varano F, Vigiani E, et al. 4-Heteroaryl substituted amino-3, 5-dicyanopyridines as new adenosine receptor ligands: novel insights on structure-activity relationships and perspectives[J]. Pharmaceuticals, 2022, 15(4): 478. https://doi.org/10.3390/ph15040478.
- Heitel P. Emerging TACnology: heterobifunctional small molecule inducers of targeted posttranslational protein modifications[J]. Molecules, 2023, 28(2): 690. https://doi.org/10.3390/molecules28020690.
- Troup R I, Fallan C, Baud M G J. Current strategies for the design of PROTAC linkers: a critical review[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 273. https://doi.org/10.37349/etat.2020.00018.
