As the field of targeted protein degradation expands, molecular glues are gaining traction as the next-generation therapeutic strategy following PROTACs. Unlike bifunctional PROTAC molecules, molecular glues work by inducing protein-protein interactions that lead to selective degradation of target proteins. In this article, we explore how molecular glues differ from PROTACs, their unique advantages, and their growing potential in tackling previously "undruggable" targets across disease areas.
What Are Molecular Glues?
Molecular glue (MG) compounds are a distinct class of small molecules that, through the degradation, stabilization or activation of the target protein post-binging, have the ability to alter protein–protein interactions (PPIs) and interactomes. Small-molecule MGs are progressively being discovered and are increasingly being recognized for their therapeutic potential for human diseases, including cancer. Multiple studies show that these small-molecule MG compounds can theoretically interact with any protein which acts as central drug targets for human diseases although some of these protein targets were once considered undruggable. Highly effective MG compounds for cancer treatment act on multiple key proteins and one key protein target can be controlled by different MG compounds with various chemical compositions. The high promiscuity of MG–protein interaction profiles has laid a fertile ground for the discovery and development of small-molecule MG compounds that can serve as molecular tools to help to dissect disease mechanisms, and to facilitate drug development for the treatment of human disease, particularly human cancer.
Defining Molecular Glues and Their Mechanism of Action
MGs comprise a category of small monovalent molecules under 500 Da which exert their function by binding to a groove or surface on one protein and reshaping its structure to create or strengthen a high-affinity interaction with another unrelated protein. As such, they are not conventional enzyme inhibitors, but rather allosteric modulators that stabilize transient or non-existent PPIs. The prototypical outcome of molecular glue action is the formation of a ternary complex—Glue: Protein A : Protein B—that can lead to two separate pharmacological consequences: functional modulation, by tuning enzymatic activity, scaffolding or sub-cellular localization; and targeted protein degradation, by recruiting an E3 ubiquitin ligase to a neo-substrate, triggering poly-ubiquitination and proteasomal degradation. The glue begins by binding to a "donor" protein (in most cases an E3 ligase such as cereblon (CRBN), VHL, or DCAF15). This event is accompanied by subtle, yet crucial, conformational changes that expose a previously cryptic hydrophobic patch or pocket of charged residues. The complementary surface on the "acceptor" protein (the disease-relevant target) is then bound with micromolar-to-nanomolar affinity, resulting in the cooperative formation of a ternary complex, whose stability is thermodynamically driven by enthalpy–entropy compensation. Structural studies show that the glue molecule commonly intercalates between α-helices or β-sheets, to act as a molecular staple that locks the PPI in place. Critically, the catalytic nature of the process means that one glue molecule can potentiate the ubiquitination and destruction of several target proteins, amplifying its PD response. This mechanism has been leveraged to target transcription factors (IKZF1/3), kinases (CK1α), and aggregation-prone proteins (tau) that were historically deemed "undruggable".
How Molecular Glues Differ from PROTACs?
Despite utilizing the same UPS machinery, molecular glues and PROTACs are structurally distinct and differ in design, mechanism, and pharmacology.
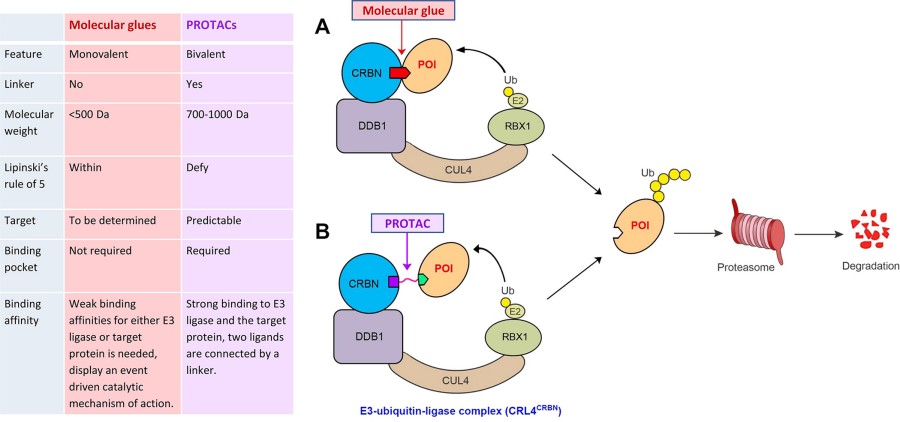 Fig. 1 Schematic presentation of the degradation of a protein of interest (POI) via the ubiquitin (Ub)–proteasome system using (A) a molecular glue or (B) a PROTAC bound to the E3 ubiquitin ligase CUL4–RBX1–DDB1–CRBN (CRL4CRBN) complex.1,2
Fig. 1 Schematic presentation of the degradation of a protein of interest (POI) via the ubiquitin (Ub)–proteasome system using (A) a molecular glue or (B) a PROTAC bound to the E3 ubiquitin ligase CUL4–RBX1–DDB1–CRBN (CRL4CRBN) complex.1,2
On a mechanistic level, MGs function via induced fit, i.e. they bind to one protein, form a new interface de novo and thereby "glue" the previously non-interacting partner to it. PROTACs, in contrast, function via proximity induction, i.e. a bifunctional molecule binds the POI and an E3 ligase simultaneously to form a forced ternary complex whose geometry is defined by linker length and flexibility. As a result, PROTACs can be designed in a modular fashion by exchanging ligands and linkers, while molecular glues must be identified or optimized through iterative screening and structural validation. The monovalent nature of molecular glues results in superior oral bioavailability, lower immunogenicity, and better blood–brain barrier penetration, desirable properties for drug candidates, especially in CNS indications. PROTACs, despite being larger molecules, exhibit exquisite target selectivity and allow for the simultaneous pursuit of multiple POIs using the same E3 ligase ligand scaffold. Interestingly, some PROTACs can be "deconstructed" into molecular glues by truncating the linker and subsequently optimizing the resulting warhead, highlighting a convergent continuum between the two modalities.
The Rise of Molecular Glues in Drug Discovery
Applications of Molecular Glues in Targeted Protein Degradation
The development of MGs has transitioned from happenstance oddities to molecularly defined scaffolds for the re-programming of the UPS to degrade therapeutic targets. In contrast to PROTACs, which necessitate two high-affinity ligands connected by a linker, molecular glues are monovalent small molecules (generally < 500 Da) that remodel the surface of an E3 ligase or its substrate to form a de novo protein–protein interface. The sparse molecular topology of molecular glues facilitates degradation of targets once thought "undruggable" due to their lack of catalytic pockets, including transcription factors, scaffolding proteins, and intrinsically disordered domains. The prototypical scenario involves the glue binding a "donor" protein, most commonly the cereblon (CRBN) adaptor of the CUL4–RBX1–DDB1–CRBN (CRL4) E3 ligase complex, that rearranges the tertiary structure of the ligase to expose a cryptic hydrophobic patch. A "client" or neo-substrate is then recruited to this patch, yielding a ternary complex whose stability is governed by positive cooperativity (ΔΔG ≈ 2–4 kcal/mol). The client is poly-ubiquitinated and trafficked to the 26S proteasome for degradation; as the glue remains intact, a single molecule can target hundreds of copies of the client, epitomizing event-driven pharmacology. The last decade has witnessed diverse applications in oncology, neurology, and immunology. In multiple myeloma, the IMiDs thalidomide, lenalidomide and pomalidomide serve as CRBN glues for the degradation of transcription factors IKZF1 and IKZF3, culminating in the suppression of IRF4 and MYC signalling. Outside of haematology, the aryl-sulfonamide indisulam glues RBM39 to DCAF15, inducing dysregulated splicing and apoptosis in lung and colorectal cancers. Cyclin K is targeted by CDK12-directed glues such as CR8 and dCeMM2–4, representing synthetic-lethal strategies in cancers with amplification of cyclin-E. The translation-termination factor GSPT1 is selectively degraded by CC-90009 (eragidomide) in relapsed/refractory acute myeloid leukaemia (AML), the first rationally designed glue to enter Phase I clinical trials. In neurodegeneration, BRD4-targeting glues suppress tau hyper-phosphorylation in murine models of Alzheimer's disease, while FKBP12-directed glues promote α-synuclein clearance in Parkinsonian organoids. These examples highlight the diversity of molecular glue utility and the potential to tackle pathogenic proteins across varied cellular compartments.
Early Successes and Examples of Molecular Glues
Historically, the most important discovery of a molecular glue began with the mechanistic explanation of thalidomide, a sedative introduced in 1957 which later caused birth defects by degradation of the developmental transcription factor SALL4. In 2010, it was shown that thalidomide binds CRBN within the CRL4 complex, thereby causing a neomorphic interface that recruits IKZF1/3 for ubiquitination. This led to the renaissance of the drug from a warning example from the past into the progenitor of a new class of immunomodulatory imide drugs (IMiDs), of which lenalidomide and pomalidomide are currently yielding > US $12 billion in annual oncology sales. A second landmark discovery from cell-based phenotypic screening campaigns was the dCeMM series (dCeMM1–4). dCeMM1 was discovered from a 2,000-compound cytotoxicity screen and shown to degrade RBM39 through bridging to DCAF15, while dCeMM2–4 were subsequently optimised to degrade cyclin K through CDK12/DDB1 engagement. These small molecules proved that a rational approach to library construction could substitute for serendipity as a discovery engine.
Comparison Between PROTACs and Molecular Glues
Similarities and Key Differences
PROTACs and MGs both belong to the emerging field of targeted protein degradation (TPD). The general aim of TPD is to ablate pathogenic proteins via the ubiquitin–proteasome system. The two strategies are connected mechanistically by the generation of a ternary complex that juxtaposes a POI with an E3 ubiquitin ligase, leading to its poly-ubiquitination and proteasomal degradation. However, the structural and operational differences between the two approaches are significant and underlie their divergent drug-discovery pathways. Glues are monovalent, single-component molecules (MW < 500 Da) that attach to either the E3 or the POI to activate a de novo protein–protein interaction (PPI). PROTACs are bivalent, hetero-bifunctional entities (MW 700–1,200 Da) that contain an independent POI ligand, an E3 ligand, and a spacer/linker. Glues are usually in accord with Lipinski's rules, resulting in better oral bioavailability, membrane permeability and brain penetration. PROTACs are more likely to break multiple Ro5 parameters, which creates challenges in formulation and delivery in vivo. Glues do not require a pre-existing high-affinity binding pocket on the POI; instead, the interaction between the E3 ligase (or the POI) is remodelled to generate a neo-interface. PROTACs require, at a minimum, moderate-affinity ligands for the POI and the E3 ligase, which makes target selection more predictable. Structural data also show that molecular glues exhibit high positive cooperativity (ΔΔG ≈ 2–4 kcal mol-1) after the PPI is nucleated but can be quite promiscuous in their selectivity, and have historically been found serendipitously. In contrast, selectivity in PROTACs is achieved through geometric restrictions by the linker, and through ligand specificity that can be tuned.
When to Choose PROTACs Over Molecular Glues (and Vice Versa)?
A characterised moderate-to-high affinity ligand is known for the POI and an E3 ligase. This allows quick, rational PROTAC construction and the establishment of predictable structure–activity relationships. PROTACs can be designed for allele- or isoform-specific degradation, as for ARV-110 to target AR mutants in prostate cancer. The same E3 ligand can be coupled to different POI ligands from combinatorial libraries, allowing rapid target-hopping across oncology, inflammation and virology programmes. PROTACs can circumvent active-site mutations that abolish inhibitor binding while retaining the ability to degrade a protein, as with BTK-directed degraders. Transcription factors (e.g., MYC, STAT3) or scaffolding proteins that have no druggable pockets can be targeted for degradation if a glue reshapes the E3 ligase surface to accommodate them. Glues' low molecular weight and favourable physicochemical profile also enables blood–brain barrier penetration, making them attractive for neurodegenerative diseases such as Alzheimer's and Huntington's. The lack of a linker simplifies synthesis, reduces the purification burden and the cost-of-goods, and speeds lead optimisation timelines. Phenotypic or chemoproteomic screens that identify previously unknown PPIs can be quickly developed if the hit compound already acts as a glue, as with indisulam-mediated RBM39 degradation.
Advantages of Molecular Glues in Drug Discovery
Higher Target Specificity and Reduced Off-Target Effects
MGs reach unparalleled target specificity, enabled by their event-driven, proximity-based mode of action in comparison to competitive occupancy of an active site. After glue engagement to form the ternary complex of an E3 ubiquitin ligase and its neo-substrate, only those proteins capable of simultaneously meeting the requirements of the newly established interface are ubiquitinated; all other proteins in the cell are "invisible" to the ligase. This geometric and electrostatic selectivity is operationalized as sub-stoichiometric catalysis, whereby a single glue molecule can be used to destroy hundreds of copies of the pathogenic protein. This obviates the need for supraphysiological doses and correspondingly lowers the chances of off-target effects. Quantitative proteomics analysis shows that lenalidomide targets IKZF1/3 for degradation in multiple myeloma with >95 % specificity and little change to the transcriptome, indicating minimal collateral signalling disruption. The small molecular footprint of glues (MW usually < 500 Da) is also well within Lipinski's rule-of-five, conferring higher membrane permeability, oral bioavailability and blood–brain barrier penetration. These physicochemical properties in turn afford lower plasma exposures for equivalent efficacy, narrowing the therapeutic window in which off-target liabilities can present. Moreover, covalent molecular glues, exemplified by JQ1 analogues that template covalent modification at the DCAF16–BRD4 interface, irreversibly lock the PPI and prevent dissociation of the subunits, thus eliminating the transient, non-productive association and dissociation events that can seed off-target ubiquitination. Altogether, these properties enable molecular glues to discriminate between highly homologous protein isoforms, rendering them a highly desirable format for allele-specific targeting in cancer and monogenic diseases.
Potential to Target "Undruggable" Proteins
Canonical drug-discovery requires that a target have a deep, ligandable pocket to host a small-molecule inhibitor. This tenet leaves out an estimated 85 % of the human proteome including transcription factors, scaffolding proteins, and aggregation-prone intrinsically disordered proteins (IDPs) . Molecular glues obviate this bottleneck by re-wiring protein–protein interactions instead of binding to the target itself. Glues form a physical bridge between an E3 ligase (such as CRBN, DCAF15, or DDB1) and a neo-substrate that has no recognizable ligand-binding cavity, forcing ubiquitination of proteins that are typically "undruggable." The success of this strategy is best illustrated by lenalidomide and pomalidomide, which degrade the transcription factors IKZF1 and IKZF3 in multiple myeloma despite these transcription factors lacking druggable pockets. More recently, high-throughput multiplexed platforms have been developed to screen thousands of compounds against purified human E3 ligases and >20,000 full-length proteins, thereby facilitating rapid identification of glues that eliminate oncogenic STAT3, β-catenin mutants, and aggregation-prone tau in models of neurodegeneration. In the case of covalent glues, such as JQ1-derived electrophiles, these compounds were shown to template the ubiquitination of BRD4—a BET family transcriptional co-activator that was previously deemed "undruggable"—by forming a covalent linkage within the DCAF16–BRD4 interface. In silico ternary-complex predictors have emerged that integrate AlphaFold2 structures with machine-learning scoring functions to enable rational glue design for otherwise intractable transcription factors such as MYC and p53 mutants that have historically been missed by small-molecule screens. Collectively, molecular glues are opening the druggable proteome to new targets beyond the traditional realm of enzymology and providing transformative new avenues for precision medicines against diseases that are driven by "undruggable" proteins.
How Our Solutions Support Molecular Glue Development?
As the field of molecular glue degraders continues to advance, researchers face new challenges in identification, validation, and optimization of glue-based therapeutics. At BOC Sciences, we provide specialized tools and services to help you stay at the forefront of this evolving protein degradation strategy. Whether you are working on early-stage screening or late-stage lead optimization, our integrated support accelerates your molecular glue discovery pipeline from idea to in vivo validation.
High-Efficiency Screening Tools for Molecular Glues
Identifying effective molecular glues requires robust, sensitive, and high-throughput-compatible screening platforms. Our screening solutions are specifically designed to address the unique pharmacological mechanisms of molecular glues, including:
- Target-agnostic screening platforms using proteomics and phenotypic assays to uncover novel glue candidates without requiring predefined binding pockets.
- Covalent and non-covalent binder profiling to assess potential molecular glue interactions with E3 ligases or substrates.
- Ternary complex stabilization assays to validate glue-induced proximity between target proteins and E3 ligases.
- Degradation efficacy readouts using real-time reporter systems and quantitative proteomics.
All our screening tools are optimized for reproducibility, scalability, and integration with medicinal chemistry workflows—ideal for both biotech startups and large pharma R&D.
Tailored Services for Molecular Glue Discovery and Optimization
Every molecular glue project is unique—and so are our service packages. We offer customized discovery and optimization programs tailored to your specific targets, therapeutic area, and stage of development. Our services include:
- Rational molecular glue design using AI-driven structure-based modeling and ligand-E3 docking prediction.
- Custom synthesis of novel small molecules, including privileged scaffolds known to enhance glue activity.
- In vitro and cell-based validation, including CRISPR-based knockdowns, degrader efficacy profiling, and target engagement studies.
- Lead optimization support, including ADME profiling, solubility improvement, and off-target minimization strategies.
By combining our deep expertise in targeted protein degradation with industry-proven technical platforms, we empower your team to go from concept to clinical candidate—faster and with greater confidence.
Interested in developing molecular glues or exploring hybrid approaches with PROTACs? We offer custom screening assays, structural design services, and validated compounds for fast-track discovery. Reach out now to learn how we can support your next breakthrough.
References
- Image retrieved from Figure 1 " Schematic presentation of the degradation of a protein of interest (POI) via the ubiquitin (Ub)–proteasome system using (A) a molecular glue or (B) a PROTAC bound to the E3 ubiquitin ligase CUL4–RBX1–DDB1–CRBN (CRL4CRBN) complex," Sasso J M.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Sasso J M, Tenchov R, Wang D S, et al. Molecular glues: the adhesive connecting targeted protein degradation to the clinic[J]. Biochemistry, 2022, 62(3): 601-623. https://doi.org/10.1021/acs.biochem.2c00245.
- Li F, Aljahdali I A M, Ling X. Molecular glues: capable protein-binding small molecules that can change protein–protein interactions and interactomes for the potential treatment of human cancer and neurodegenerative diseases[J]. International Journal of Molecular Sciences, 2022, 23(11): 6206. https://doi.org/10.3390/ijms23116206.
- Kubryń N, Fijałkowski Ł, Nowaczyk J, et al.PROTAC Technology as a New Tool for Modern Pharmacotherapy[J]. Molecules, 2025, 30(10): 2123. https://doi.org/10.3390/molecules30102123.
- Han X, Sun Y. PROTACs: A novel strategy for cancer drug discovery and development[J]. MedComm, 2023, 4(3): e290. https://doi.org/10.1002/mco2.290.
