The development of PROTACs (PROteolysis TArgeting Chimeras) has transformed modern drug discovery by enabling selective protein degradation rather than inhibition. Since their first conceptualization in the early 2000s, PROTACs have evolved into a major class of therapeutic candidates with broad applications in oncology, neurology, and immunology. In this article, we trace the history, technological milestones, and ongoing advancements that have shaped the rise of PROTACs in biomedical research and pharmaceutical development.
The Birth of PROTAC Technology
The PROTAC was first reported in 2001, and PROTAC technology has advanced rapidly in the past 20 years. PROTAC degraders have now entered clinical Phase I-II, with more than 10 molecules, and ARV-471 and ARV-110 from Arvinas, Inc are the most advanced PROTACs for the treatment of recurrent breast cancer and prostate cancer, respectively. However, PROTAC is still confronted with many challenges.
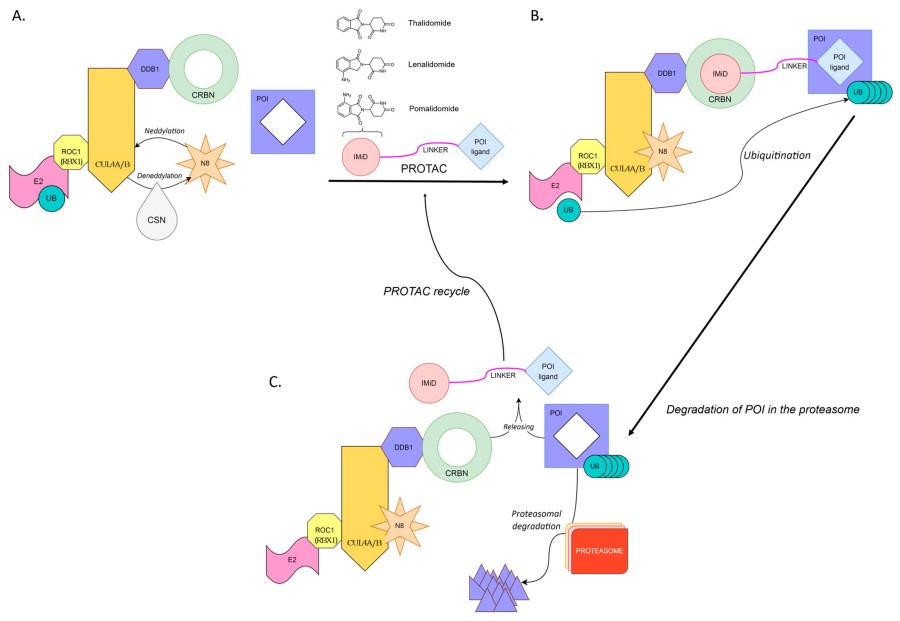 Fig. 1 PROTAC-induced degradation of a target protein.1,2
Fig. 1 PROTAC-induced degradation of a target protein.1,2
Early Discoveries and Breakthroughs in Protein Degradation
While the foundational concepts that underlie TPD date to the late 1990s, the first mention of what would now be called PROTAC technology was in a 1999 patent application from the biotech company Proteinix, in which "chimeric small molecules" that harness the ubiquitin–proteasome system (UPS) for therapeutic purposes were described. The UPS had long been known to be the major pathway for selective protein degradation inside cells: a ubiquitin-conjugation cascade (E1 → E2 → E3) ultimately adds poly-ubiquitin chains to lysine residues on substrates, which tags the substrates for 26S proteasomal degradation. The key step that was still missing was a universal chemical approach to harness this endogenous protein quality-control system to target a protein of interest (POI). In 2001, Crews and Deshaies delivered this missing proof-of-concept. Their first PROTAC, called "Protac-1", was a 10-mer phosphopeptide that simultaneously bound the F-box protein β-TRCP (an adaptor for E3 ligases) and methionine aminopeptidase-2 (MetAP-2), and promoted MetAP-2 ubiquitination and degradation in Xenopus egg extracts. Critically, Protac-1 established the event-driven logic: degradation is catalytic, and one PROTAC can mediate the destruction of multiple MetAP-2 molecules. In this report, the term "PROTAC" (PROteolysis-TArgeting Chimera) was coined, and the modular design (POI ligand + linker + E3 ligand) that is still in use today was established.
Key Milestones in PROTAC Research
The first PROTAC was published by the Crews and Deshaies laboratory in 2001, which employed the SCFb-TRCP E3 to target methionine ami-nopeptidase-2 (MetAp-2) for degradation. Crews' laboratory replaced the peptidic MDM2 ligand with the bona fide small molecule Nutlin-3 to generate an AR-targeting PROTAC in 2008, which maintained cell permeability and mediated degradation in the micromolar range. This seminal advance set in motion an industry-wide shift towards drug-like chemical matter. High-resolution crystal structures of VHL in complex with hydroxyproline-containing ligands were published by the Ciulli laboratory, allowing for the rational design of small, high-affinity VHL mimetics with improved physicochemical properties. These VHL ligands rapidly became the standard building blocks for second-generation PROTACs. Two near simultaneous reports established thalidomide analogues as CRBN ligands, which led to the development of the first nanomolar-potency BET-PROTACs (dBET1, MZ1). With the emergence of potent, orally bioavailable E3 ligase ligands, PROTAC construction was democratised, which led to an explosion of pre-clinical publications (>200 in 2015–2017). ARV-110, an AR-targeting degrader, became the first PROTAC to show oral efficacy in murine xenograft models, and the door was opened for human trials. ARV-110 and ARV-471 (ER degrader) entered Phase I trials, officially transitioning PROTACs from academic tool to investigational therapeutic. Crews' laboratory published TRAFTACs in 2021, a CRISPR-anchored approach that exploits recruitment of dCas9-fused HaloTag to guide PROTACs to endogenous transcription factors such as MYC, taking advantage of, and further blurring the line between, genetic and pharmacological modalities. PROTACs targeting IRAK4 (inflammatory disease), α-synuclein (Parkinson's), and mutant huntingtin (Huntington's) entered Phase I trials, signalling the maturation of the platform across therapeutic areas.
The Evolution of PROTACs in Drug Discovery
Early Applications of PROTACs in Targeted Therapy
Conceptual precursors to PROTAC technology first emerged in 2001, when Sakamoto and colleagues used a 10-mer phosphopeptide conjugate (later called "Protac-1") to redirect the Skp1–Cullin–F-box (SCF) ubiquitin-ligase complex to methionine aminopeptidase-2 (MetAP-2) in Xenopus egg extracts. Although micromolar in potency and poorly cell-permeable, this peptide chimera validated the event-driven approach: a single bifunctional ligand can induce poly-ubiquitination and proteasomal degradation of a protein target, obviating the need for sustained active-site occupancy. The second wave of development arrived in 2004 when Crews' laboratory tethered a cell-penetrating HIV-TAT peptide to a hypoxia-inducible factor (HIF-1α) VHL-binding domain, creating a first cell-penetrant construct capable of inducing the degradation of the androgen receptor (AR) and estrogen receptor (ER). These early seminal efforts expanded the druggable proteome into transcription factors and scaffolding proteins, and thus presented the potential for intervention in hormone-driven cancers. Independently, a handful of academic laboratories followed suit by leveraging "molecular glues" (e.g. thalidomide derivatives) that target the cereblon (CRBN) E3 ligase to target transcription factors IKZF1 and IKZF3 for degradation, presenting the first blueprints to repurpose FDA-approved drugs as degrader scaffolds. The first small-molecule PROTAC, published in 2008, swapped out the peptidic VHL ligand for Nutlin-3 (a MDM2 binder), and demonstrated oral bioavailability in mice xenograft models, showing that modular medicinal-chemistry optimisation could mitigate the liabilities of peptide chemistry . By 2013, phosphoPROTACs—peptide-based degraders whose E3-recruiting motif becomes phosphorylated only in the presence of oncogenic signalling—afforded the first in vivo demonstration of tumour growth inhibition, validating the concept of context-dependent degradation . Taken together, these early successes transformed PROTACs from academic curiosities into candidate therapeutics with the potential to circumvent mechanisms of resistance that confound conventional kinase inhibitors and hormone antagonists.
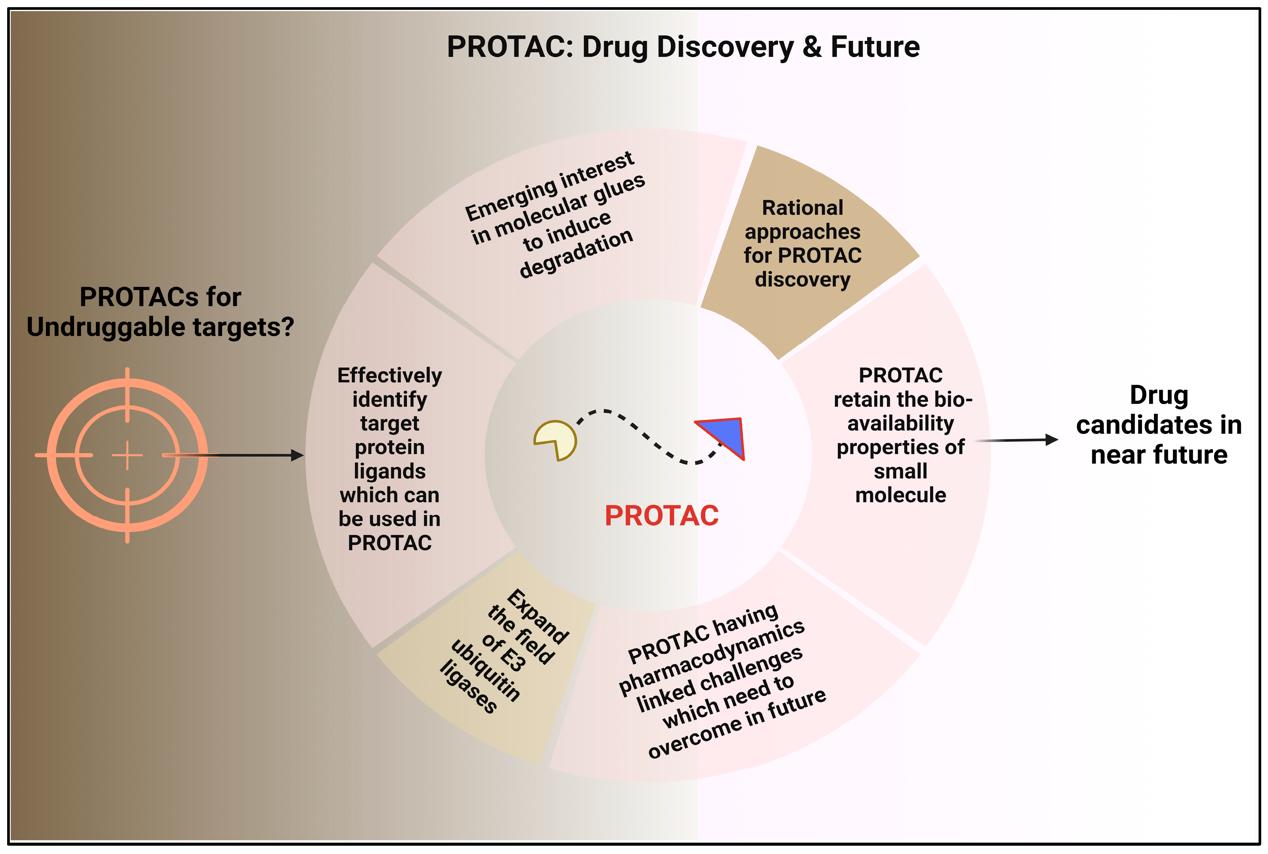 Fig. 2 The Future of Drug Discovery with PROTAC Technology.3,4
Fig. 2 The Future of Drug Discovery with PROTAC Technology.3,4
Major Research Institutions and PROTAC Innovations
Key academic and industrial drivers have further accelerated the pace of PROTAC development. Yale University (Crews Laboratory) continues to be a hub for basic discovery and chemical development; early contributions include the first peptide-based PROTAC-1 and the first orally bioavailable degrader ARV-110, which advanced to Phase I clinical trials in 2019. The University of Dundee (Ciulli Laboratory) led structure-guided efforts to elucidate high-resolution crystal structures of VHL–PROTAC–POI ternary complexes. These structures enabled rational optimisation of linker length and stereochemistry, facilitating >100-fold improvements in cellular potency. Kyoto University (Hashimoto Laboratory) developed a new platform of "bioPROTACs" that leverage non-canonical amino acids to achieve photochemical control, enabling light-activated PHOTACs that spatiotemporally control the degradation of BRD4 or HDAC6. Dana-Farber Cancer Institute and the Broad Institute have employed CRISPR dropout screens, in combination with quantitative proteomics, to nominate synthetic-lethal PROTAC targets in TP53-mutant or RB-deficient cancers. In industry, Arvinas Therapeutics (New Haven, CT), spun out of Yale in 2013, has been instrumental in translating basic PROTAC principles into clinical-stage degraders: ARV-110 (AR) and ARV-471 (ER) have collectively enrolled >500 patients in Phase I–III clinical trials, and established proof-of-concept for oral PROTAC pharmacokinetics and safety. C4 Therapeutics and Kymera Therapeutics have identified potent ligands for DCAF16 and RNF114, respectively, thereby expanding the diversity of E3 ligase platforms, which could complement existing CRBN-dependent PROTACs with orthogonal degradation modalities and reduced CRBN-related immunomodulatory liabilities. In 2023, a collaborative consortium between Oxford, Genentech, and Boehringer Ingelheim has opened a new avenue of PROTAC-enabled PROTACs (PEPs), where a first degrader potentiates the action of a second degrader to create sequential cascades that result in ultra-rapid target clearance.
PROTAC Development Pathways
Design and Screening of PROTAC Molecules
PROTAC discovery has developed into a multi-parameter optimisation exercise that connects three pharmacophores, i.e. target ligand, E3-recruiting ligand, and a flexible linker, into a single catalytic unit. Typically, ligand identification for both the protein-of-interest (POI) and E3 ligase is initiated with high-throughput fragment screening (HTFS), DNA-encoded libraries (DELs), or in-silico docking against AlphaFold2-predicted structures to identify hundreds of potential binders. After "warheads" are validated by SPR or nano-DSC, linker design is started. Linker length, rigidity, and hydrophilicity are then varied (often 10–20 atoms) to optimise cooperativity within the ternary complex; cryo-EM snapshots of PROTAC–POI–E3 assemblies are now being used to direct these decisions with near-atomic resolution. Medicinal-chemistry cycles in parallel incorporate structure–activity relationship (SAR) matrices that weigh potency against drug-like properties. Early-stage property-based design now enables liabilities like cLogP > 7 or topological polar surface area (tPSA) > 180 Å2 to be eliminated before synthesis, speeding up hit-to-lead development. Screening cascades have also advanced beyond simple western blotting to include luminescent HiBiT tags, NanoBRET live-cell degradation assays, and even multiplexed proteomics (TMT-MS), enabling 384-well quantification of on-target degradation and even global off-target signatures within 48 h. To interrogate catalytic efficiency, a "hook-effect" counter-screen is performed at 0.01–10 µM to identify a concentration window in which ternary complex formation is favoured over binary artifacts. Machine-learning models trained on >5,000 PROTAC datapoints can now predict DC, D, and off-target risk, saving around 40 % of experimental iterations. Finally, parallel synthesis of regio-isomer-controlled libraries, combined with DoE-guided optimisation, has pushed solid-phase yields above 60 % and lowered impurity levels to<1 %, streamlining the transition into pre-clinical PK/PD evaluation.
Overcoming Developmental Hurdles
The molecular elegance of PROTACs is compromised by their trilemma of ADME, selectivity, and manufacturing. Pharmacokinetics is challenged by high molecular weight (800–1,200 Da), low passive permeability and extensive hepatic metabolism by CYP3A4/UGT1A1. Remedies include prodrug masking of polar handles by bioreversible esters to improve oral bioavailability from<5 to="">30 %, linker rigidification to reduce the entropy loss and associated efflux liability, and salt-form and nano-milling approaches to increase kinetic solubility >200 µg/mL, a necessary criterion for immediate-release tablets. Selectivity liabilities emanate from a limited repertoire of E3 ligases (mostly CRBN, VHL, MDM2 and IAPs) and from neomorphic "neo-substrate" degradation (e.g. GSPT1 or IKZF1/3). The ligase toolbox is now being expanded by DEL selection against TRIM21, RNF43 and FEM1B, while CRISPR-based profiling of E3 expression is used for patient stratification to ligase-high tumours. Manufacturing hurdles arise from regio-isomer formation (up to 70 %), low convergent yields (<35 %) and scale-dependent aggregation; these are overcome by flow-chemistry protocols, DoE-optimised one-pot conjugations and QbD-controlled crystallisation to reproducibly deliver kilogram-scale GMP batches.
Current State of PROTAC Research and Industry Trends
PROTACs in Cancer and Other Diseases
By mid-2025 the global PROTAC pipeline has disclosed more than 120 programmes that include over 71 oncology-focused projects and 12 active human trials. Breast and prostate carcinomas remain predominant in late-stage development, consistent with the clinical head start accrued by the ER and AR degraders. Vepdegestrant (ARV-471) has advanced into two pivotal Phase III trials. VERITAC-2 will directly compare vepdegestrant monotherapy with fulvestrant in endocrine-refractory ER/HER2 metastatic breast cancer, while VERITAC-3 is exploring the combination with palbociclib in the first-line setting. Promising signals in early phases have not yet translated into convincing efficacy in expansion cohorts, with 38 % clinical benefit rate despite 3 % objective response rate. Going forward, selection of biomarker-enriched cohorts will be critical for success. Bavdegalutamide (ARV-110) and ARV-766 are in Phase II in metastatic castration-resistant prostate cancer, with ARV-110 eliciting ≥50 % PSA decline in ~40 % of patients harbouring AR ligand-binding-domain mutations. Clinical expansion is accelerating beyond the canonical steroid receptors. DT2216 (Dialectic), a VHL-recruiting BCL-XL degrader, is accruing patients with relapsed T-cell lymphoma and small-cell lung cancer in a Phase I dose-escalation study. FHD-609 (Foghorn) and CFT-8634 (C4 Therapeutics) are exploring BRD9 degradation in synovial sarcoma, while KT-333 has recently been granted orphan-drug designation for STAT3-driven peripheral T-cell lymphoma. Pre-clinical programmes are now targeting KRAS-driven pancreatic cancer (LHF418 SOS1 degrader), BRAF melanoma (CFT-1946), and cholangiocarcinoma (ARV-825 BRD4 degrader), each achieving sub-nanomolar DC values and robust tumour regression in xenograft models.
Growing Industry Interest in PROTAC Technologies
The market landscape for targeted protein degradation has matured rapidly since 2020. Funding across the space has topped USD 4.8 billion in venture capital between 2021-2024, while the number of pre-clinical PROTACs has quadrupled over the same period. Arvinas, a spin-out from Yale University, maintains its first-mover status with 2 Phase III assets (ARV-471, ARV-110) and a fully-licensed CRBN ligand platform that Pfizer acquired for up to USD 2.1 billion. C4 Therapeutics is building on its TORPEDO degron-engineering platform to propel CFT-1946 (BRAF) and CFT-8634 (BRD9) into Phase I/II, in partnership with Roche and Biogen respectively. Kymera Therapeutics has developed a heterogeneous pipeline including IRAK4, STAT3 and SMARCA2 degraders, with KT-474 earning Fast Track designation for hidradenitis suppurativa and atopic dermatitis. Nurix Therapeutics is on the cusp of submitting an IND for NX-5948, a next-generation BTK degrader with activity against the C481S mutant resistant form, and able to completely suppress BTK in cynomolgus monkeys at 3 mg kg. Contract research organisations (CROs) have also entered the fray, with some offering "one-stop shops" for PROTAC services – DEL screening, cryo-EM ternary-complex validation, GLP toxicology, etc – accelerating timelines from 36 months down to 18 months. Big-pharma engagement has been just as voracious: Novartis, AstraZeneca and Johnson & Johnson have each set up joint ventures worth more than USD 1 billion each with a focus on transcription-factor degraders for solid tumours. Regulatory momentum is also beginning to snowball: the FDA has already granted 4 Fast Track and 2 Breakthrough Therapy designations to PROTAC candidates since 2022, a sign of faith in the risk-benefit of the modality. In sum, strong pre-clinical efficacy, growing target diversity and an influx of capital and collaborations suggest that PROTAC technology will be a mainstay of 21st-century pharmacotherapy.
Why Choose Us for PROTAC Development?
With the rapid rise of PROTAC technology as a transformative approach in drug discovery, working with a knowledgeable and agile partner is crucial for success. At BOC Sciences, we specialize in targeted protein degradation and offer end-to-end support for PROTAC development-from early design and synthesis to in vitro and in vivo validation. Whether you're developing your first degrader or advancing a lead compound toward clinical trials, our tailored solutions help you achieve your goals faster and more efficiently.
Our Expertise in PROTAC Research and Development
We have been deeply involved in the evolution of PROTACs from academic concept to translational application. Our multidisciplinary team brings together expertise in medicinal chemistry, structural biology, bioconjugation, and cell-based assays. Our capabilities include:
- Rational design of PROTAC molecules based on known ligands and E3 ligase binding preferences.
- Extensive experience with common E3 ligases, including CRBN, VHL, MDM2, and IAP.
- Optimization of linker chemistry to improve cellular permeability, stability, and ternary complex formation.
- Integration of AI-assisted modeling for structure-activity prediction and degrader design.
- Real-time project communication and support throughout every research milestone.
By combining domain expertise with a client-focused approach, we ensure your PROTAC R&D stays aligned with both scientific rigor and commercial timelines.
Comprehensive PROTAC Design Services to Fast-Track Your Research
Our service platform is designed to accelerate your PROTAC discovery process while minimizing cost and development risk. Whether you're starting from a validated target or a novel concept, our offerings include:
- Custom PROTAC synthesis with a wide range of warheads, linkers, and E3 ligase ligands.
- Linker scanning libraries to rapidly identify optimal linkers for your target-ligase pair.
- Cell-based degradation assays to evaluate efficacy, selectivity, and cellular response.
- Pharmacokinetic (PK) and ADMET profiling to support compound progression.
- Scalable manufacturing support for preclinical studies and early development batches.
With our one-stop PROTAC development workflow, you gain a reliable partner capable of transforming your protein degradation strategy into a viable therapeutic program. Let us help you shorten your development timeline and increase your probability of success in the clinic.
Stay ahead in the evolving world of targeted protein degradation. Our team provides expert PROTAC design support, screening platforms, and tailor-made molecule synthesis to streamline your R&D. Contact us for high-performance solutions that drive innovation.
References
- Image retrieved from Figure 1 " PROTAC-induced degradation of a target protein," Cieślak M.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Cieślak M, Słowianek M. Cereblon-recruiting PROTACs: Will new drugs have to face old challenges?[J]. Pharmaceutics, 2023, 15(3): 812. https://doi.org/10.3390/pharmaceutics15030812.
- Image retrieved from Figure 8 " The Future of Drug Discovery with PROTAC Technology," Sincere N I.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Sincere N I, Anand K, Ashique S, et al. PROTACs: emerging targeted protein degradation approaches for advanced druggable strategies[J]. Molecules, 2023, 28(10): 4014. https://doi.org/10.3390/molecules28104014.
- He M, Cao C, Ni Z, et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021)[J]. Signal transduction and targeted therapy, 2022, 7(1): 181. https://doi.org/10.1038/s41392-022-00999-9.
- Yedla P, Babalghith A O, Andra V V, et al. PROTACs in the management of prostate cancer[J]. Molecules, 2023, 28(9): 3698. https://doi.org/10.3390/molecules28093698.
- Kubryń N, Fijałkowski Ł, Nowaczyk J, et al. PROTAC Technology as a New Tool for Modern Pharmacotherapy[J]. Molecules, 2025, 30(10): 2123. https://doi.org/10.3390/molecules30102123.
