With the rise of targeted protein degradation, PROTACs are increasingly being compared to traditional small molecule inhibitors in terms of mechanism, efficacy, and drug resistance profiles. This article offers a detailed comparison of PROTAC degraders and conventional inhibitors, examining the pros and cons of each approach. If you're considering a shift toward degrader-based therapeutics, understanding the fundamental differences between these modalities is essential.
The Traditional Approach: Small Molecule Inhibitors
Small molecule inhibitors (SMIs) have been one of the most useful types of drugs as well as key chemical tools to study the biology of the target protein of interest. The recent development in diverse -omics technologies has accelerated the rate of identifying the disease-relevant proteins. However, these need to be translated into human use through the development of selective small molecule chemicals to inhibit the target proteins. The classical small molecule inhibitors, which function through binding to either orthosteric sites or allosteric sites of the target protein, are suffering from several challenges, including drug selectivity, therapy resistance as well as drugging undruggable and multi-domain proteins. PROTACs have been suggested as a potential solution. PROTACs are heterobifunctional molecules with a binding ligand for a protein of interest and an E3 ligase-recruiting ligand that are linked together via a chemical linker. Upon binding to the target protein, PROTAC brings a E3 ligase close enough for it to be initiated to polyubiquitinate the target protein, and thus for its proteasome-mediated degradation. PROTACs work to degrade the target protein entirely, and do so in a catalytical way, in contrast to small molecule inhibitors.
How Small Molecule Inhibitors Work?
SMIs are small-molecular-weight (< 900 Da) molecules designed to diffuse into cells, bind to a target protein with high affinity, and inhibit its biochemical activity. Mechanistically, they can be classified into reversible and irreversible modes of action. Reversible inhibitors occupy the active or allosteric site of an enzyme or receptor, thereby sterically precluding substrate binding or conformational activation. Binding is mediated by non-covalent interactions (hydrogen bonds, π-stacking, van der Waals contacts, and salt bridges) whose free energy of binding (ΔG) is sufficient to shift the equilibrium toward the inactive state. Irreversible inhibitors, by contrast, form covalent bonds with catalytic residues—e.g., ebselen forms a selenyl-sulfide linkage with Cys145 of SARS-CoV-2 3CL, permanently disabling the viral protease. Structural biology has elucidated two canonical binding paradigms: competitive and allosteric. Competitive inhibitors recapitulate the transition state or substrate geometry, as exemplified by T863 binding within the acyl-CoA tunnel of DGAT1, thereby occluding triacylglycerol synthesis. Allosteric inhibitors stabilise inactive enzyme conformations by binding to distal regulatory pockets; this mechanism underpins the action of the DGAT1 inhibitor DGAT1N1, which locks the membrane-embedded lateral gate of DGAT1 in a closed state. In signalling cascades, kinase inhibitors such as imatinib bind to the ATP-binding cleft of BCR-ABL, stabilising an inactive "DFG-out" conformation and halting downstream oncogenic signalling . Overall, the exquisite complementarity between inhibitor and binding pocket provides nanomolar potency and, in many cases, isoform selectivity.
Key Success Stories and Limitations of Small Molecule Inhibitors
SMIs have made landmark successes in the clinic and the markets. Imatinib (Gleevec) changed the treatment of chronic myeloid leukaemia (CML) by inducing long-lasting remissions in > 90 % of newly diagnosed BCR-ABL-positive patients. Lenalidomide and pomalidomide are transforming agents in the treatment of multiple myeloma (MM), which target IKZF1/3 transcription factors, and have generated > US $12 billion of annual sales. Ritonavir-boosted lopinavir is a strong antiviral in HIV and SARS-CoV-2, the DHODH inhibitor BAY-2402234 has been reported to be potent and well tolerated in COVID-19 pre-clinical and early-phase clinical studies.
On the other hand, the clinical use of small molecular inhibitors is also faced with some intrinsic obstacles. First, the target proteins of small molecular inhibitors are typically enzymes and receptors with pockets or active sites. Roughly 75% of the human proteome is active-site-less (transcription factors, scaffolding proteins, and non-enzymatic proteins), which are in turn undruggable by this strategy. Second, in order to provide an adequate intracellular concentration to the target proteins for therapeutic effects, relatively high systemic drug levels need to be sustained. However, because the small molecular inhibitors are highly competitive, they are often associated with off-target effects and adverse side effects. Third, a typical small molecule typically only abolishes the function of one domain of multidomain scaffolding proteins. The functional activities of other domains in the scaffold and their interactions with other proteins are not affected. In cancer cells, the inhibition of multidomain kinases can also induce feedback compensation to activate their downstream signaling pathways through alternative kinases. For instance, treatment of cells with bromodomain inhibitors of multidomain and bromo-containing proteins, such as TRIM24, were not able to display effective anti-proliferative response, which suggests that bromodomain inhibition alone is not a successful anti-cancer strategy. Fourth, protein overexpression and protein accumulation from induced compensatory protein expression may lead to incomplete depletion of the target proteins and incomplete inhibition of downstream signaling pathways.
Advantages of PROTACs Over Small Molecule Inhibitors
Heterobifunctional PROTAC molecules are composed of a ligand to target protein, a ligand to E3 ubiquitin ligase and a linker region linking the two ligands. After target: PROTAC:E3 ternary complex formation, ubiquitin is transferred by E2 ubiquitin-conjugating enzymes to lysine residues on the surface of the target protein. Recognition of lysine polyubiquitination signal by the proteasome leads to the degradation of the target protein. A number of small-molecule PROTACs with favorable pharmaceutical properties have been reported in recent years.
Targeted Protein Degradation and Broader Application
Conventional SMIs follow an occupancy-driven pharmacology. They must bind with high fractional occupancy to an active or allosteric site in order to exert pharmacologic activity. This has limited the druggable proteome to only the 15–20 % of proteins in the human genome that have well-ordered ligand-binding pockets. PROTACs (proteolysis-targeting chimeras), by contrast, follow an event-driven pharmacology as catalytic degraders. The bifunctional PROTAC molecule transiently forms a ternary complex with a POI of interest, even if the POI is enzymatically inert, and an E3 ubiquitin ligase. When poly-ubiquitin chains are added, the POI is targeted for destruction in the 26S proteasome and the PROTAC molecule is recycled to catalyze the next reaction. This catalytic activity has several important consequences. First, lower (sub-stoichiometric) doses of PROTAC are needed to deplete a target because PROTACs themselves are not degraded. Nanomolar concentrations of PROTACs are sufficient to deplete micromolar concentrations of target protein. As a result, PROTACs achieve their effect with lower systemic exposure, and thus less off-target liabilities. Second, because degradation abolishes the entire polypeptide chain including scaffolding/adaptor domains and non-catalytic functions, PROTACs silence all functional outputs of a target protein, and not just enzymatic activity. This has made it possible to target traditionally "undruggable" proteins including transcription factors (STAT3, MYC), structural proteins and aggregation-prone proteins. Third, PROTACs can be re-engineered to work in other subcellular compartments using lysosome-targeting chimeras (LYTACs) or nano-PROTACs, which have been used to degrade membrane receptors and extracellular proteins, such as EGFR or PD-L1, inaccessible to SMIs. Fourth, PROTACs can be tuned to be isoform-selective by exploiting subtle differences in the surfaces of related proteins, a property rare for traditional inhibitors. Overall, these properties make PROTACs a versatile, pan-proteomic platform to treat a wide variety of protein-associated diseases, including oncology, inflammation, neurodegeneration, and even viral infection.
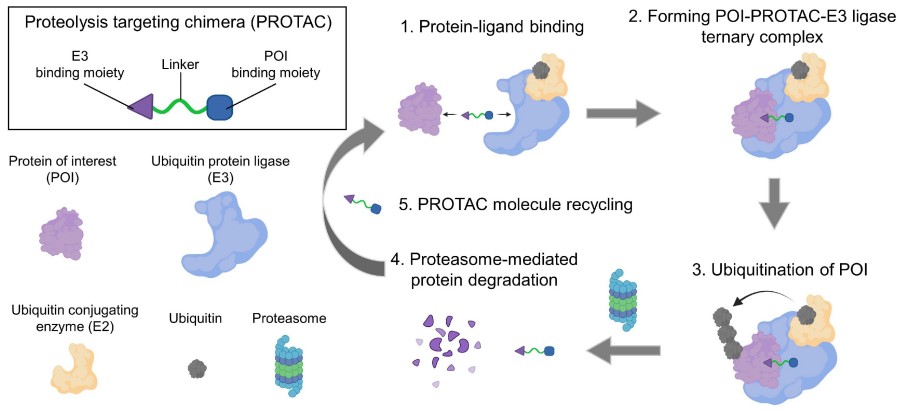 Fig. 1 Schematic illustration of the mechanism of PROTAC.1,2
Fig. 1 Schematic illustration of the mechanism of PROTAC.1,2
Overcoming Drug Resistance with PROTACs
Resistance to SMIs has been well characterized, and is known to emerge from a limited set of 3 main, convergent mechanisms: target point mutations in the active site that weaken drug binding, overexpression of the target, or activation of compensatory pathways. PROTACs avoid each of these resistance mechanisms. Since PROTACs eliminate the entire protein, not just its catalytic function, they cannot be bypassed by a change in function. PROTACs also do not need to bind with high affinity to the catalytic site itself; weak or allosteric ligands are still potent if they can induce a ternary complex. As a result, gate-keeper mutations or kinase domain mutations that confer resistance to ATP-competitive SMIs do not affect PROTACs. (ABL, EGFR, BTK etc.) The mutation does not affect the ubiquitination machinery so even mutant proteins are targeted. For example ARV-110 (see table) degrades androgen receptor (AR) with L702H and H875Y mutations, conferring enzalutamide resistance, and resulted in ≥50 % PSA decline in Phase II patients. Amplification of a target is also circumvented, since the drug continues to degrade the protein at a faster rate than it can be produced. This has been demonstrated in EGFR-amplified non-small-cell lung cancer (NSCLC) xenografts using EGFR-directed nano-PROTACs. In addition, if cancer cells evolved to down-regulate the E3 ligase recruited by an original PROTAC, an alternate E3 ligase can be rationally re-engineered (e.g., CRBN, VHL, IAPs) This re-engineering is also a resistance-resistant approach. Studies in pre-clinical models show PROTACs that degrades the oncogenic driver and its compensatory signalling node (e.g. CDK4/6 and PI3K) pre-empt the cell's adaptive rewiring, something that is not possible with traditional single-site SMIs.
Challenges and Opportunities in PROTAC Development
In stark contrast to this extraordinary progress in the clinic (>25 degraders in human trials), a principled approach to PROTAC design is still hampered by a set of mutually reinforcing molecular and safety bottlenecks. First, physicochemical liabilities: most leads are >800 Da and have TPSA >150 Ų, leading to poor aqueous solubility, low intestinal permeability, and highly variable oral bioavailability. Recent structure-based efforts have sought to reverse these liabilities through linker truncation, non-polar isosteric replacement and pro-drug masking; but to date, only a few "beyond-Ro5" degraders have attained meaningful exposures. Second, E3-ligase selectivity and off-target degradation: the current clinical arsenal is heavily biased toward CRBN and VHL, both of which are widely expressed and therefore pose a greater threat of degrading critical housekeeping proteins. Promising approaches to address this issue include exploring the remaining >600 human E3 ligases for those with a restricted tumour expression (e.g. DCAF15, FEM1B), as well as CRISPR dropout screens to identify ligases that are least essential. Third, on-target toxicity: whereas inhibitors only transiently bind to their target, PROTACs effectively abolish target expression, a much greater insult to cellular proteostasis. Although the catalytic mode of action and sub-stoichiometric dosing should in theory spare natural substrates, the high Cmax levels needed to degrade rapidly-turnover proteins has been linked to sustained depletion of HIF-1α and associated cardiopulmonary toxicities in pre-clinical studies. Safety pharmacology cascades are increasingly leveraged with multiplexed proteomics, single-cell RNA-seq, and organoid-on-chip assays to flag liabilities prior to first in human dosing.
Why PROTACs Could Replace Small Molecule Inhibitors in the Future?
The Potential of PROTACs to Revolutionize Drug Discovery
PROTACs are rapidly evolving from a scientific curiosity into a platform of choice for therapeutic intervention, with the potential to replace many traditional small-molecule inhibitors. The primary benefit of these PROTACs is their event-driven, catalytic mechanism of action: a PROTAC does not directly occupy an active site, but rather recruits an E3 ubiquitin ligase to label the protein of interest for proteasomal degradation, and is then freed to catalyse subsequent degradation cycles. This confers a catalytic advantage, as it allows for sub-stoichiometric dosing and therefore reduced systemic exposure, off-target toxicity, and dosing frequency, all of which are major limitations of occupancy-based drugs. In addition, PROTACs greatly expand the druggable proteome from occupancy-based small molecules by targeting not only classical proteins with hydrophobic binding pockets, but also transcription factors (e.g., STAT3, MYC), scaffolding proteins, and aggregation-prone, intrinsically disordered proteins that have been historically resistant to small-molecule development. Algorithmic and structural development have also led to greatly increased discovery efficiency: machine-learning guided de-novo design software such as AiPROTAC can now predict ternary-complex (TCE) stability and in vivo degradation potency entirely in silico, reducing iterative medicinal-chemistry efforts from months to days. In addition, next-generation technologies such as dual-target PROTACs, peptide-based PROTACs, CLIPTACs, and PHOTACs, enable spatiotemporal control, oral availability, and tissue-selectivity not possible with traditional occupancy-based inhibitors without extensive formulation development. These advantages are likely to place PROTACs at the forefront of the next generation of drug discovery platforms to overcome the high failure rates and resistance liabilities of small molecules.
Real-World Applications: Case Studies
ARV-110 led to a ≥50 % PSA decline in ≈40 % of mCRPC patients harboring ligand-binding-domain mutations (AR-L702H, H875Y) that confer complete resistance to enzalutamide or abiraterone in Phase II trials . The PROTAC selectively degraded both wild-type and mutant AR and showed mutation-agnostic efficacy unattainable by traditional antagonists. Two concurrent Phase III trials (VERITAC-2 and VERITAC-3) are evaluating ARV-471 monotherapy or combination with palbociclib in ER/HER2 metastatic breast cancer. Preliminary results show 38 % clinical benefit rate and 62 % tumor ER knock-down, validating PROTAC superiority in overcoming endocrine resistance. In BL cell lines, the small-molecule BET inhibitor JQ1 stabilizes BRD4 and induces compensatory c-MYC up-regulation. ARV-825, a CRBN-recruiting PROTAC, rapidly and irreversibly depletes BRD4, thereby suppressing MYC transcription and inducing >90 % apoptosis—a result unobtainable by JQ1 alone.
Our Expertise in PROTAC vs. Small Molecule Research
With over a decade of hands-on experience in targeted protein degradation and small molecule drug discovery, we offer a unique cross-disciplinary perspective that empowers pharmaceutical and biotech companies to choose the most effective therapeutic strategy. Whether you're exploring PROTAC technology or continuing development with traditional small molecule inhibitors, our scientific team and lab infrastructure are fully equipped to support you at every stage.
High-Performance Screening for PROTACs and Small Molecule Inhibitors
We provide comprehensive, high-throughput screening platforms specifically designed to accommodate the distinct mechanisms of PROTACs and traditional inhibitors. Our capabilities include:
- Cell-based degradation assays for rapid evaluation of PROTAC-mediated target knockdown.
- Biochemical binding assays for small molecule potency and selectivity profiling.
- Dual-platform compound libraries containing validated scaffolds for both PROTAC and inhibitor development.
- Advanced analytics and proteomics to assess functional outcomes.
- Real-time degradation kinetics and dose-response profiling under physiologically relevant conditions.
These tools ensure that your compound's efficacy, selectivity, and pharmacodynamics are characterized with the precision required for high-stakes decision-making in preclinical research.
Solutions Tailored to Your Research Needs
We understand that choosing between a PROTAC strategy and a traditional small molecule inhibitor depends on multiple factors—such as target accessibility, resistance mechanisms, and therapeutic objectives. That's why we offer flexible, customized service packages that align with your unique R&D goals:
- Target assessment consultations to help determine whether degradation or inhibition is more appropriate.
- PROTAC rational design and linker optimization based on E3 ligase compatibility and target topology.
- Medicinal chemistry services for inhibitor hit-to-lead development and SAR (structure-activity relationship) exploration.
- Integrated in vitro & in vivo studies to support data-driven progression from screening to IND-enabling studies.
Whether you're launching a new discovery program or pivoting from inhibition to degradation, we provide the scientific insight and technical firepower to help you make informed, confident choices.
Ready to transition from inhibitors to degraders? We offer end-to-end PROTAC development solutions—from initial design and synthesis to in vitro evaluation. Talk to our experts and discover how our tools and technologies can elevate your drug discovery program.
References
- Image retrieved from Figure 1 " Schematic illustration of the mechanism of PROTAC," Liu Z.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Liu Z, Zhang Y, Xiang Y, et al. Small-molecule PROTACs for cancer immunotherapy[J]. Molecules, 2022, 27(17): 5439. https://doi.org/10.3390/molecules27175439.
- Zhao L, Li S, Zhong W. Mechanism of action of small-molecule agents in ongoing clinical trials for SARS-CoV-2: a review[J]. Frontiers in Pharmacology, 2022, 13: 840639. https://doi.org/10.3389/fphar.2022.840639.
- Ouyang M, Feng Y, Chen H, et al. Recent Advances in Optically Controlled PROTAC[J]. Bioengineering, 2023, 10(12): 1368. https://doi.org/10.3390/bioengineering10121368.
- Liu Z, Hu M, Yang Y, et al. An overview of PROTACs: a promising drug discovery paradigm[J]. Molecular biomedicine, 2022, 3(1): 46. https://doi.org/10.1186/s43556-022-00112-0.
