RIPTAC Promises to Overcome Cancer Treatment Resistance
* Please be kindly noted that our services and products can only be used for research to organizations or companies and not intended for any clinical or individuals.
Consult with Our Experts
Article
Cancer treatment resistance remains one of the most complex challenges in oncology research. Even when a cancer model initially responds to a targeted compound, resistant cell populations can emerge through target mutation, pathway rewiring, adaptive transcriptional programs, altered protein expression, and tumor heterogeneity. These mechanisms can reduce the long-term value of strategies that depend on blocking a single oncogenic driver or one signaling node.
Regulated induced proximity targeting chimeras, widely known as RIPTACs, introduce a different way to think about selective cancer cell targeting. Instead of relying only on direct inhibition of a cancer-driving protein, a RIPTAC molecule is designed to bring together a tumor-associated target protein and an essential protein. When the target protein is enriched in a resistant cancer cell context, the RIPTAC can induce a TP:RIPTAC:EP ternary complex and restrict essential protein function selectively in target-expressing cells. This "hold-and-kill" concept is attracting attention because it may open new research strategies for cancer models where classical target inhibition becomes insufficient.
Why Cancer Treatment Resistance Remains a Major Research Challenge?
Cancer resistance is not a single event. It is often a dynamic process involving genetic changes, phenotypic plasticity, altered protein networks, and selective pressure from repeated compound exposure. In many research models, sensitive cells may be eliminated first, while subpopulations with alternative survival routes become more dominant over time. This complexity makes resistance difficult to address with a single inhibitory mechanism. For researchers developing next-generation targeted strategies, the central question is not only whether a compound can block a known oncogenic signal. It is also whether the strategy can remain effective when the cell changes the target, bypasses the pathway, or shifts into a resistant state.
Target Mutations That Reduce Drug Sensitivity
Target mutation is one of the most widely studied mechanisms of cancer treatment resistance. A small change in the binding site, conformation, or regulatory region of a protein may reduce compound binding or alter downstream signaling behavior. In kinase-focused research, for example, mutations can change the shape of an inhibitor-binding pocket, stabilize an active conformation, or weaken the interaction between the compound and its intended target. Mutation-driven resistance creates an important design problem. If a compound relies on a precise binding mode to inhibit a catalytic function, even a subtle structural alteration can reduce activity. Researchers may then need to design next-generation inhibitors, explore alternative binding pockets, or identify a new vulnerability in the resistant cell. RIPTAC offers an additional conceptual route: the target protein may serve as a selective recruitment handle if it remains present in resistant cells, even when its signaling behavior has changed.
Pathway Bypass and Adaptive Signaling Networks
Many cancer cells can survive pathway inhibition by activating alternative signaling circuits. A blocked pathway may be replaced by parallel growth factor signaling, compensatory transcriptional programs, altered feedback loops, or changes in survival protein expression. This type of adaptive network behavior can make direct pathway inhibition less durable in resistant cell models. Bypass signaling is especially challenging because it does not always require loss of the original target. The target may remain expressed and even engaged by the compound, while the cell uses another route to maintain proliferation, stress tolerance, or survival. In this context, a RIPTAC strategy may be valuable because it can potentially transform a persistently expressed cancer-associated protein into a functional proximity anchor, creating a vulnerability that is not limited to the original signaling pathway.
Tumor Heterogeneity and Resistant Cell Subpopulations
Tumor heterogeneity means that cancer cell populations are not uniform. Different subpopulations may vary in gene expression, protein abundance, metabolic state, cell-cycle status, stress response, and compound sensitivity. Within the same experimental model, one group of cells may remain sensitive to pathway inhibition, while another group survives through altered signaling or a different cellular state. This heterogeneity complicates target selection. A target that is important in one subpopulation may be less relevant in another. For RIPTAC research, this creates both a challenge and an opportunity. The challenge is to identify target proteins that are sufficiently enriched across resistant cell states. The opportunity is to exploit expression patterns that distinguish resistant cancer cells from target-low cells, even when those target proteins are not classical oncogenic drivers.
Limitations of Targeting Only Classical Oncogenic Drivers
Traditional targeted strategies often prioritize proteins that directly drive cancer cell growth or survival. This approach has generated many useful research tools, but it can also narrow the target space. Some resistant cancer cells may not depend on one dominant driver. Others may retain driver expression but escape through non-genetic adaptation, altered protein networks, or redundant signaling. RIPTAC research expands the way target proteins can be viewed. A target protein does not always need to be the central driver of a cancer model. It may instead function as a selective cellular address, guiding the RIPTAC molecule into a target-expressing cell state and helping recruit an essential protein into a ternary complex. This distinction is important for resistance research because it allows researchers to consider proteins that were previously less attractive for direct inhibition but remain valuable as selective recognition elements.
Table 1. Key resistance mechanisms and RIPTAC-relevant research opportunities.
| Resistance mechanism | Research challenge | RIPTAC-relevant opportunity |
|---|
| Target mutation | Reduced sensitivity to compounds designed for a specific binding mode | Use persistent target expression as a selective proximity anchor |
| Pathway bypass | Alternative signaling maintains survival despite pathway inhibition | Create a functional vulnerability through essential protein sequestration |
| Tumor heterogeneity | Mixed cell states respond differently to the same compound | Design molecules around target-enriched resistant subpopulations |
| Non-driver dependence | Some resistant cells do not rely on one dominant oncogenic driver | Leverage non-driver proteins with selective expression profiles |
What Is RIPTAC?
RIPTAC stands for regulated induced proximity targeting chimera. It is a heterobifunctional small-molecule concept designed to bring two proteins into close proximity: a target protein that is selectively or highly expressed in a cancer cell context, and an essential protein that supports important cellular functions. Through this induced proximity event, the RIPTAC molecule promotes formation of a ternary complex and restricts essential protein function in target-expressing cells. The key idea is selective functional interference. Rather than degrading the target protein or simply blocking its active site, RIPTAC uses the target protein as a molecular address. Once the RIPTAC binds the target protein and recruits an essential protein, the resulting complex can reduce the essential protein's functional availability. This mechanism may selectively affect cells where the target protein is sufficiently expressed, making it attractive for resistance research based on differential protein expression.
Definition of Regulated Induced Proximity Targeting Chimeras
A regulated induced proximity targeting chimera is a designed molecule that induces a controlled interaction between two proteins that would not otherwise form the same productive complex under normal conditions. In RIPTAC research, these two proteins are commonly described as the target protein, or TP, and the essential protein, or EP. The RIPTAC bridges them together and stabilizes a TP:RIPTAC:EP ternary complex. The word "regulated" is important because the activity of the molecule depends on protein expression and complex formation. If the target protein is abundant in the cell, the RIPTAC has more opportunity to form the intended ternary complex. If the target protein is absent or expressed at low levels, the complex is less likely to accumulate. This expression-dependent behavior is one reason RIPTACs are discussed as a potential strategy for selective activity in resistant cancer models.
Core Components of a RIPTAC Molecule
A RIPTAC molecule is typically designed around three functional components. The first is a ligand that binds the target protein. This ligand determines which target-expressing cell context the molecule may engage. The second is a ligand that binds the essential protein. This component determines which essential protein function may be sequestered or restricted after ternary complex formation. The third is a linker that connects the two ligands and controls the distance, orientation, flexibility, and physicochemical behavior of the whole molecule. Although this architecture looks conceptually simple, the design requirements are highly complex. Strong binary binding to each protein is not always enough. The molecule must position the target protein and essential protein in a geometry that supports productive protein-protein proximity. Linker length, exit vector, polarity, rigidity, conformational flexibility, and cellular exposure can all influence whether a RIPTAC candidate forms a stable and functional ternary complex.
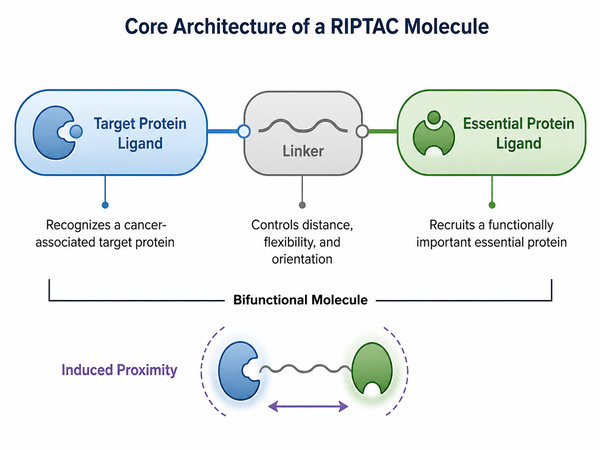 Fig.1 Core architecture of a RIPTAC molecule (BOC Sciences Original).
Fig.1 Core architecture of a RIPTAC molecule (BOC Sciences Original).
Why RIPTAC Is Gaining Attention in Cancer Resistance Research?
RIPTAC is gaining attention because it offers a way to explore cancer cell selectivity beyond the traditional requirement of inhibiting a disease-driving protein. In resistant cancer models, the protein that best identifies the resistant cell state may not be the best protein to inhibit. RIPTAC separates these roles. The target protein can serve as a recognition anchor, while the essential protein becomes the functional vulnerability. This separation creates a broader design logic. Researchers can search for proteins that are enriched in resistant cells, evaluate whether they are ligandable, and then test whether they can support productive recruitment of an essential protein. If successful, this strategy may help address resistance states driven by adaptive signaling, heterogeneous protein expression, or incomplete dependence on classical oncogenic drivers.
How RIPTAC Works in Resistant Cancer Cells?
The action of a RIPTAC molecule can be understood as a sequence of linked events. First, the molecule recognizes a target protein that is enriched in the cancer cell model. Second, it recruits an essential protein through the other binding arm. Third, the target protein, RIPTAC molecule, and essential protein form a ternary complex. Fourth, the essential protein's normal function is restricted in a target-dependent manner, leading to selective cellular effects in the target-expressing population.
Recognizing Cancer-Associated Target Proteins
Target protein recognition is the first determinant of RIPTAC selectivity. A useful target protein should show a meaningful expression difference between the resistant cancer model and the comparison cell system. It should also have a ligandable site or a binding interface that can support small-molecule engagement. In some projects, the target protein may be a receptor, transcription-associated protein, kinase, epigenetic regulator, or other intracellular protein with disease-relevant expression. For resistance research, the most useful target is not always the most famous oncogenic driver. A target protein may be valuable because it marks a resistant state, remains expressed after treatment pressure, or distinguishes a cell subpopulation that survives conventional inhibition. This makes expression profiling, proteomic analysis, and target validation important early steps in RIPTAC discovery.
Recruiting Essential Proteins Through Induced Proximity
The essential protein component gives RIPTAC its functional impact. Essential proteins participate in cellular processes required for survival, proliferation, transcriptional maintenance, mitotic progression, or other core functions. A RIPTAC molecule does not need to eliminate the essential protein from the cell. Instead, it can restrict its functional availability by holding it in a ternary complex with the target protein. Essential protein recruitment must be carefully designed. If the interaction is too weak, the ternary complex may not persist long enough to produce a measurable cellular response. If the essential protein ligand is too broadly active outside the ternary complex context, selectivity may be reduced. The research goal is to generate a molecule whose most meaningful activity depends on target protein-mediated proximity.
Forming a Stable TP:RIPTAC:EP Ternary Complex
The TP:RIPTAC:EP ternary complex is the central mechanistic event in RIPTAC activity. A candidate molecule may bind both proteins separately, but still fail if it cannot bring them into a productive orientation. Therefore, ternary complex formation, stability, and cooperativity are often more informative than binary affinity alone. Cooperativity describes how the binding of one protein influences recruitment of the second protein. Positive cooperativity can stabilize the ternary complex and improve cellular activity. Negative cooperativity can weaken complex formation even when both binary interactions appear strong. For this reason, RIPTAC design should evaluate the entire three-part system rather than optimizing each binding arm in isolation.
Disrupting Essential Protein Function Through the "Hold-and-Kill" Mechanism
The "hold-and-kill" concept describes the functional logic of RIPTAC. The molecule holds the target protein and essential protein together in a stable complex. This proximity event restricts the essential protein's normal role in the target-expressing cancer cell, creating a selective cellular effect. The target protein acts like a location-dependent anchor, while the essential protein provides the functional vulnerability. For resistant cancer cell models, this mechanism may be especially useful when the original driver pathway is no longer sufficient as a single intervention point. Instead of trying only to re-inhibit a mutated or bypassed pathway, RIPTAC research asks whether the resistant cell's protein expression pattern can be used to create a new proximity-based vulnerability.
How RIPTAC May Overcome Cancer Treatment Resistance?
RIPTAC may help overcome cancer treatment resistance by changing the rules of target selection. Traditional strategies often ask whether a protein is the disease driver and whether direct inhibition of that protein can suppress cancer cell growth. RIPTAC research asks an additional question: can a protein that is selectively expressed in resistant cancer cells be used to recruit and restrict an essential protein? This difference matters because resistance often reduces the effectiveness of single-pathway inhibition. If a resistant cell remains identifiable through a target-enriched protein, a RIPTAC molecule may convert that protein into a functional anchor. This strategy can potentially address resistance mechanisms that arise from target mutation, bypass signaling, heterogeneous cell populations, or adaptive cell states.
Targeting Resistant Cells Without Relying Solely on Driver Inhibition
Driver inhibition depends on the continued dominance of a disease-driving protein or pathway. In resistant cancer models, that dominance may be weakened by mutation, compensation, or network rewiring. A RIPTAC molecule does not necessarily require the target protein to remain the main driver of survival. It requires the target protein to be sufficiently expressed and accessible for ligand binding. This creates a research opportunity for proteins that are difficult to inhibit directly or are not ideal standalone targets. If such proteins are enriched in resistant cells, they may still be useful as recognition elements. By recruiting an essential protein into a target-dependent ternary complex, RIPTAC design can turn expression selectivity into functional selectivity.
Using Differential Protein Expression to Improve Cellular Selectivity
Differential protein expression is a central concept in RIPTAC research. A target protein that is highly expressed in resistant cancer cells but low in comparison cells may provide a selectivity window. The RIPTAC molecule can accumulate or act more effectively where the target protein is present, enabling selective formation of the TP:RIPTAC:EP complex. To evaluate this possibility, researchers should compare target protein expression across sensitive cells, resistant cells, target-low control cells, and relevant disease models. Quantitative protein measurement, immunodetection, flow cytometry, cell imaging, and proteomic profiling can help determine whether the selected target has a useful expression pattern for RIPTAC design.
Turning Tumor-Associated Proteins into Functional Vulnerability Anchors
In many research programs, tumor-associated proteins are evaluated as direct inhibition targets. RIPTAC introduces another use: the same protein may serve as an anchor that brings an essential protein into a restricted state. This is especially meaningful when the tumor-associated protein is abundant and ligandable, but its direct inhibition is not sufficient to overcome resistance. The anchor concept expands the usable target space. A protein may be valuable if it is selectively expressed, intracellularly accessible, and able to support ternary complex formation. It does not always need to have a catalytic pocket, a known driver function, or a direct relationship to the resistant phenotype. The key is whether its presence can regulate the proximity-induced effect.
Restricting Essential Protein Activity in Target-Expressing Cancer Cells
Essential proteins are usually difficult to target selectively because they are broadly important across many cell types. RIPTAC design seeks to solve this problem by making essential protein restriction dependent on target protein expression. The essential protein is not simply inhibited everywhere. Instead, its functional interference is linked to the formation of a ternary complex in target-expressing cells. This target-dependent restriction is why ternary complex analysis is so important. Researchers must determine whether essential protein engagement occurs primarily in the intended proximity context. They should also compare cellular activity between target-positive and target-low systems to determine whether the observed effect is truly dependent on the selected target protein.
Addressing Adaptive and Heterogeneous Resistance Mechanisms
Adaptive resistance can involve broad changes in cell state. Heterogeneous resistance can involve multiple subpopulations with different survival routes. RIPTAC may be useful in these settings if a shared target protein remains expressed across resistant populations or if a specific target marks a difficult-to-eliminate resistant subgroup. For research design, this means RIPTAC candidates should be tested in models that reflect resistance diversity. Single-cell profiling, resistant cell line panels, co-culture systems, and target expression stratification can help determine whether a RIPTAC molecule has activity across heterogeneous populations or only within a defined target-positive subset.
Key Advantages of RIPTAC for Cancer Resistance Research
The main value of RIPTAC lies in its ability to connect selective protein expression with functional essential protein restriction. This makes it different from strategies that rely only on target inhibition, pathway blockade, or target protein removal. For cancer resistance research, the advantages are especially relevant when resistant cells remain target-identifiable but are no longer fully dependent on one inhibited pathway.
Expanding Targeting Opportunities Beyond Conventional Druggable Proteins
Conventional druggability often focuses on proteins with well-defined active sites, pockets, or catalytic functions. Many proteins that are relevant to resistant cell states do not fit this model. They may be scaffolding proteins, transcription-associated proteins, or proteins with surfaces that are difficult to inhibit directly. RIPTAC research can expand the targetable space by using a protein as a recognition anchor rather than a direct inhibition target. A ligand with sufficient binding and selectivity may be useful even if it does not strongly suppress the target protein's intrinsic function. This broadens the types of proteins that can be considered in resistance-focused discovery programs.
Leveraging Non-Driver Proteins for Selective Cancer Cell Targeting
Non-driver proteins are often overlooked when the goal is direct pathway inhibition. However, some non-driver proteins may show strong differential expression in resistant cancer cells. If they are intracellularly accessible and ligandable, they may provide a selective entry point for RIPTAC design. This is an important conceptual shift. A protein can be useful because it identifies the resistant cell, not because it independently drives the resistant phenotype. RIPTAC converts this recognition value into functional activity by recruiting an essential protein into a target-dependent complex.
Reducing Dependence on Single-Pathway Inhibition
Single-pathway inhibition can be vulnerable to feedback activation and pathway redundancy. RIPTAC does not eliminate the need to understand pathways, but it offers another layer of intervention. By restricting an essential protein through induced proximity, RIPTAC can create a cellular consequence that is not limited to blocking one upstream signaling node. This may be valuable in resistant models where multiple pathways contribute to survival. Instead of chasing every bypass route individually, researchers can evaluate whether a stable target protein anchor can direct essential protein restriction in the resistant cell population.
Supporting New Strategies for Resistant and Heterogeneous Cancer Models
Resistant and heterogeneous cancer models require strategies that can be adapted to different target expression patterns. RIPTAC is modular by design. Researchers can modify the target protein ligand, essential protein ligand, or linker to explore different protein pairs and cellular contexts. This modularity supports iterative optimization. A project can begin with target expression profiling, move into ligand selection, test several linker designs, and then evaluate ternary complex formation and cellular effects across target-positive and target-low models. Each step can generate data for the next design cycle.
Providing a Flexible Platform for Induced Proximity-Based Molecule Design
Induced proximity has become an important principle in chemical biology because it allows small molecules to create new functional relationships between proteins. RIPTAC applies this principle to selective essential protein restriction in target-expressing cells. The platform is flexible because the molecular architecture can be adjusted through ligand choice, linker chemistry, and protein-pair selection. For B2B research teams, this flexibility is useful because different programs may require different design priorities. Some projects may emphasize target protein selectivity. Others may focus on ternary complex stability, cell permeability, solubility, or assay development. RIPTAC research can be customized around the biological question and the available target information.
Potential Applications of RIPTAC Technology in Cancer Research
RIPTAC technology is still an emerging research area, but its design logic is relevant to several cancer resistance contexts. The most suitable applications are likely to involve models where resistant cells show a clear target protein expression pattern, where direct driver inhibition has limitations, or where an essential protein can be functionally restricted through proximity.
Drug-Resistant Solid Tumor Models
Drug-resistant solid tumor models are a natural area for RIPTAC research because they often show heterogeneous cell states and adaptive resistance mechanisms. Resistant cells may retain expression of tumor-associated proteins even after they become less sensitive to a pathway inhibitor. These proteins may be explored as anchors for proximity-induced essential protein restriction. In a solid tumor research workflow, target expression profiling should be combined with functional assays. Researchers can compare sensitive and resistant cell models, identify proteins enriched after resistance selection, and test whether ligands against those proteins support RIPTAC-like ternary complex formation. This helps determine whether the resistant state creates a usable molecular address.
Hormone Signaling-Related Resistance Research
Hormone signaling-related resistance is often associated with altered receptor expression, receptor mutation, pathway adaptation, and transcriptional rewiring. A receptor or receptor-associated protein may remain abundant even when direct antagonism becomes less effective. This makes receptor-rich resistant models potentially relevant for RIPTAC concept exploration. In these systems, researchers can evaluate whether a hormone signaling-associated protein can serve as a target protein anchor. The goal is not limited to blocking receptor signaling directly. Instead, the target protein may help recruit an essential protein into a complex that produces selective effects in receptor-expressing cells.
Kinase Inhibitor Resistance Research
Kinase inhibitor resistance can arise through kinase domain mutation, pathway bypass, gene amplification, or activation of parallel signaling nodes. In many cases, the original kinase or associated signaling protein remains expressed in resistant cells. This persistent expression may provide an opportunity for RIPTAC-inspired design. Researchers can examine whether kinase-associated proteins in resistant models are suitable as target protein anchors. When direct inhibition becomes less durable, proximity-based functional interference may provide another research strategy. This is particularly relevant when the kinase itself is mutated but still present, or when a downstream or parallel protein better marks the resistant cell state.
Resistance Driven by Adaptive Cellular States
Some resistant cells survive not through a single stable mutation but through adaptive phenotypic states. These states may involve altered transcription, stress response, metabolic rewiring, or changes in cell-cycle behavior. Such adaptive states may be difficult to address with one classical target inhibitor because the cell can shift between survival programs. RIPTAC research can support the identification of state-associated protein anchors. If a protein is enriched in an adaptive resistant state and can be bound by a ligand, it may be used to recruit an essential protein in that specific cellular context. This application requires careful expression analysis and functional validation because adaptive states can be dynamic and reversible.
Target Validation in Cancer-Associated Protein Networks
RIPTAC also provides a useful framework for target validation. Instead of asking only whether a protein should be inhibited, researchers can ask whether the protein can guide selective proximity-induced activity. This expands target validation from functional dependency analysis to expression-based and proximity-based evaluation. Target validation for RIPTAC research should include protein abundance, localization, ligandability, expression stability, and compatibility with essential protein recruitment. A target that fails as a direct inhibitor target may still succeed as a proximity anchor if it supports a productive TP:RIPTAC:EP complex.
Need Customized RIPTAC Development Support?
BOC Sciences supports diverse RIPTAC research needs with tailored solutions covering molecular design, compound synthesis, target validation, activity evaluation, and optimization support.
Get Consultation
RIPTAC Molecular Design Considerations
RIPTAC molecular design requires integrated optimization of ligand binding, linker architecture, ternary complex formation, cellular exposure, and functional response. Because the molecule must coordinate two proteins, small structural changes can strongly influence activity. A successful design workflow should evaluate the whole system rather than treating each component as an isolated module.
Target Protein Ligand Design and Optimization
The target protein ligand controls cell-context recognition. It should bind the selected target protein with suitable affinity and selectivity while tolerating chemical modification for linker attachment. The ligand does not always need to be a potent inhibitor of the target protein, but it must support the correct orientation for ternary complex formation. Optimization should consider binding mode, exit vector, structure-activity relationship, cellular engagement, and target selectivity. If structural information is available, protein-ligand modeling can help identify attachment positions that preserve binding while allowing the linker to project toward the essential protein ligand. If structural information is limited, analog screening and binding assays can help identify ligand series suitable for RIPTAC construction.
Essential Protein Ligand Selection
The essential protein ligand determines which functional protein will be recruited into the ternary complex. This choice requires careful consideration because essential proteins are broadly important. The ligand should contribute to proximity-dependent activity without causing excessive target-independent effects. Researchers should evaluate the essential protein's cellular role, ligandability, expression level, subcellular localization, and compatibility with the target protein. The most useful essential protein partner is not necessarily the one with the strongest standalone ligand. It is the one that supports target-dependent complex formation and produces a measurable, selective cellular effect when held in the intended proximity arrangement.
Linker Length, Rigidity, Polarity, and Spatial Orientation
The linker is a central design element in RIPTAC molecules. It controls the distance between the two ligands, influences the relative orientation of the target protein and essential protein, and affects the physicochemical properties of the full molecule. Linkers that are too short may prevent productive complex formation. Linkers that are too long or too flexible may reduce cooperativity and increase conformational cost. Linker optimization should evaluate length, polarity, rigidity, hydrogen-bonding capacity, attachment chemistry, and exit vector compatibility. PEG-based, alkyl, aromatic, heteroaryl, and conformationally constrained linkers may produce very different effects on solubility, permeability, and ternary complex geometry. For this reason, linker libraries and iterative analog design are often important in RIPTAC discovery.
Ternary Complex Cooperativity and Stability
Ternary complex cooperativity and stability often determine whether a RIPTAC candidate can produce target-dependent cellular activity. A molecule may have acceptable binary affinity for both proteins but still fail to form a productive three-part complex. Conversely, a molecule with moderate binary affinity may perform well if the induced protein-protein interface is highly cooperative. Biophysical and biochemical assays can help measure complex formation, dissociation behavior, and cooperativity. Structural modeling and protein-protein interaction analysis can also support design decisions. These data help researchers understand whether activity is driven by stable proximity rather than nonspecific binding or target-independent essential protein inhibition.
Physicochemical Properties for Cellular Activity Studies
RIPTAC molecules may have higher molecular weight and greater structural complexity than many conventional small molecules. Therefore, physicochemical properties are important from the beginning of design. Solubility, chemical stability, plasma stability, permeability, aggregation tendency, and cellular exposure can all affect the interpretation of activity data. A candidate that forms a strong ternary complex in a purified system may show weak cellular activity if it cannot reach the relevant intracellular compartment. Conversely, a molecule with moderate biochemical activity may perform better in cells if it has favorable exposure and stability. Integrating physicochemical profiling with cellular assays helps avoid misleading conclusions during optimization.
Table 2. BOC Sciences services supporting RIPTAC molecular design.
Assay Strategies for Evaluating RIPTAC Activity Against Resistant Cancer Cells
RIPTAC evaluation should connect molecular binding with cellular function. A complete workflow typically includes binary binding assays, ternary complex formation analysis, cell-based functional readouts, selectivity assessment, and mechanism validation. Because the activity depends on induced proximity, no single assay is sufficient to confirm a RIPTAC mechanism.
Target Protein and Essential Protein Binding Assays
Binary binding assays are useful early in the workflow because they confirm whether each ligand arm can engage its intended protein. Common research approaches may include surface plasmon resonance, microscale thermophoresis, fluorescence polarization, isothermal titration calorimetry, enzyme-linked formats, or other biochemical and biophysical methods. For RIPTAC candidates, binding assays should evaluate both free ligands and bifunctional molecules when possible. Linker attachment can alter ligand orientation, steric fit, or binding kinetics. Measuring binding after conjugation helps determine whether the full molecule retains the required target protein and essential protein interactions.
Ternary Complex Formation and Cooperativity Analysis
Ternary complex assays are central to RIPTAC evaluation. These assays determine whether the target protein, RIPTAC molecule, and essential protein can form a stable complex. They can also reveal whether the presence of one protein improves or weakens recruitment of the other protein. Assay formats may include proximity-based fluorescence systems, time-resolved energy transfer methods, pull-down assays, native mass spectrometry, size-exclusion analysis, or other complex-detection approaches. The goal is to measure complex formation across concentration ranges, identify optimal activity windows, and detect nonproductive binding patterns.
Cell-Based Functional Activity Assays
Cell-based assays determine whether biochemical proximity translates into a measurable cellular response. These assays may evaluate cell viability, proliferation, apoptosis-related markers, transcriptional changes, pathway modulation, cell-cycle behavior, or other functional endpoints relevant to the selected essential protein. Because RIPTAC activity should depend on target protein expression, functional assays should include both target-positive and target-low cells. A selective response in target-positive cells supports the intended proximity-driven mechanism, while broad activity across all models may indicate target-independent essential protein effects or nonspecific cytotoxicity.
Resistance Model-Based Cellular Evaluation
Resistance model-based evaluation is essential for a page focused on cancer treatment resistance. Researchers should test candidate molecules in cell systems that reflect the resistance mechanism of interest, such as mutation-driven resistance, pathway bypass, adaptive signaling, or heterogeneous target expression. Useful workflows may include matched sensitive and resistant cell pairs, long-term compound-exposed models, engineered target-expression systems, and multi-cell panels with varying target protein abundance. These models help determine whether RIPTAC activity is linked to the resistant state rather than a general cellular stress response.
Selectivity Assessment Across Target-Positive and Target-Low Cells
Selectivity assessment should compare activity across target-positive, target-low, and target-negative systems. If the RIPTAC mechanism is working as intended, target expression should influence ternary complex formation and downstream cellular response. Expression knockdown, overexpression, or rescue experiments can help validate the relationship between target abundance and activity. Quantitative selectivity analysis can include EC50, maximum response, time-dependent activity, cell-state sensitivity, and target expression correlation. These data help researchers determine whether the candidate molecule is suitable for further optimization or whether its activity is too broad to support target-dependent conclusions.
Mechanism Validation for RIPTAC-Induced Cellular Effects
Mechanism validation connects the observed cellular effect to the intended RIPTAC mechanism. Researchers can evaluate whether activity depends on target protein expression, essential protein engagement, ternary complex formation, and the selected linker architecture. Competition assays using free ligands can help determine whether either binding arm is required. Additional validation may include target protein knockdown, essential protein binding competition, time-course experiments, localization studies, and orthogonal readouts of essential protein function. The strongest RIPTAC datasets connect biochemical complex formation, target-dependent cellular selectivity, and functional pathway effects in a coherent workflow.
Table 3. Suggested assay workflow for RIPTAC activity evaluation.
| Evaluation stage | Purpose | Representative readouts |
|---|
| Binary binding | Confirm engagement of target protein and essential protein | Affinity, kinetics, ligand competition, binding retention after linker attachment |
| Ternary complex analysis | Determine whether the RIPTAC bridges both proteins productively | Complex formation, stability, cooperativity, concentration-response behavior |
| Cellular activity | Measure functional response in target-expressing cells | Cell viability, proliferation, pathway readouts, apoptosis-related markers |
| Selectivity validation | Compare activity across target-positive and target-low models | Expression-response correlation, target knockdown, competition rescue |
| Resistance model testing | Evaluate activity in disease-relevant resistant cell systems | Matched cell panels, adaptive resistance models, heterogeneous population analysis |
Challenges in RIPTAC Development for Cancer Resistance Research
RIPTAC is a promising research concept, but it also presents several development challenges. The molecule must identify the right target protein, recruit a suitable essential protein, form a productive ternary complex, enter relevant cells, and produce a selective functional response. Each step can become a bottleneck if not evaluated systematically.
Identifying Suitable Protein Pairs in Resistant Cancer Contexts
Protein-pair selection is one of the most important challenges. A target protein may be enriched in resistant cells, but it may not support the correct geometry for essential protein recruitment. An essential protein may be functionally important, but its ligand may cause broad activity outside the intended ternary complex context. Researchers should begin with target expression, ligandability, localization, and essential protein compatibility. Computational modeling, protein interaction prediction, and early ternary complex assays can help prioritize protein pairs before extensive synthesis. This reduces the risk of investing heavily in combinations that are unlikely to form productive complexes.
Balancing Binding Affinity with Productive Ternary Complex Formation
Higher affinity is not always better in RIPTAC design. Very strong binding to one protein may favor binary complex formation and reduce the probability of productive ternary assembly. In other cases, weak affinity may prevent sufficient cellular engagement. The optimal balance depends on the target protein, essential protein, linker, and cellular context. Concentration-response analysis is therefore important. Researchers should evaluate whether the molecule shows a clear activity window, whether complex formation decreases at excessive concentrations, and whether cellular activity correlates with ternary complex data. This helps distinguish productive proximity from simple target binding.
Achieving Selective Activity in Heterogeneous Cell Populations
Heterogeneous cell populations complicate selectivity analysis. A candidate may show activity in a bulk culture, but the response may be driven by only one target-high subpopulation. Alternatively, target-low cells may survive and become dominant after prolonged exposure. These possibilities should be evaluated with careful experimental design. Single-cell or subpopulation-level analysis can help researchers understand whether RIPTAC activity is distributed across the resistant population or concentrated in a defined subset. Target expression stratification, imaging-based assays, and flow cytometry can provide valuable information about selectivity and resistance within heterogeneous systems.
Translating Mechanistic Insights into Optimized RIPTAC Candidates
Mechanistic insight is only useful if it can guide optimization. A weak RIPTAC candidate may fail because of poor target binding, poor essential protein recruitment, an unsuitable linker, weak cellular exposure, or insufficient ternary complex stability. Identifying the true limiting factor requires a coordinated assay strategy. Optimization should proceed through iterative design cycles. Structural modeling can guide linker modification. Binding assays can confirm ligand engagement. Ternary complex assays can evaluate cooperativity. Cell-based assays can test target-dependent activity. Physicochemical profiling can reveal solubility or stability limitations. Together, these data help convert an early concept into a more refined RIPTAC candidate.
Future Directions of RIPTAC in Cancer Treatment Resistance Research
The future of RIPTAC research will depend on better target discovery, improved ligand design, more predictive ternary complex modeling, and stronger cellular validation workflows. As researchers learn which protein pairs and molecular architectures produce selective activity, RIPTAC may become an important addition to the induced proximity toolbox for cancer resistance research.
Expanding RIPTAC Design Beyond Classical Oncogenic Drivers
One of the most important future directions is the expansion of target selection beyond classical oncogenic drivers. Resistant cancer cells may express many proteins that are not ideal direct inhibition targets but still provide useful selectivity markers. These proteins can become valuable starting points for RIPTAC design if they are ligandable and compatible with essential protein recruitment. This direction may broaden the range of research models that can be addressed by small-molecule proximity strategies. Instead of focusing only on proteins with established driver functions, researchers can evaluate expression-defined vulnerabilities and use RIPTAC architecture to create functional consequences from selective recognition.
Integrating Multi-Omics Data to Identify Resistance-Associated Targets
Multi-omics data can improve RIPTAC target discovery by connecting gene expression, protein abundance, pathway activation, and resistant cell states. Transcriptomics can identify state-associated markers, while proteomics can confirm whether those markers are present at the protein level. Phosphoproteomics and interactomics can reveal pathway adaptations and protein networks that support resistant phenotypes. For RIPTAC design, protein-level evidence is especially important. The target protein must be present in sufficient abundance and the right cellular location to support ternary complex formation. Integrating multi-omics analysis with ligandability assessment can help prioritize targets with both biological relevance and molecular design feasibility.
Combining Computational Design with Experimental Screening
Computational design can accelerate RIPTAC research by predicting ligand binding, linker geometry, protein-protein proximity, and ternary complex compatibility. Docking, molecular dynamics simulation, protein-protein interaction modeling, and structure-based design can help narrow the design space before synthesis. However, computational predictions should be validated experimentally. RIPTAC activity depends on cellular exposure, protein abundance, complex kinetics, and functional response. The most effective workflows will combine in silico modeling with biochemical assays, cellular assays, and iterative structure-activity learning.
Building End-to-End RIPTAC Research Workflows
An end-to-end RIPTAC workflow should connect target discovery, ligand design, linker optimization, synthesis, binding analysis, ternary complex evaluation, cellular testing, and mechanism validation. Each stage should generate data that informs the next stage. This integrated approach is particularly important for cancer resistance research because the biological context is complex and highly model-dependent. BOC Sciences supports researchers with a broad range of capabilities that can be adapted to RIPTAC-inspired projects, including ligand design, linker optimization, computational modeling, binding analysis, complex formation assays, cellular activity evaluation, and physicochemical profiling. These services can help research teams build a more systematic path from concept to optimized molecule.
Our Support
RIPTAC Research Support at BOC Sciences
BOC Sciences provides integrated research support for RIPTAC-inspired molecule discovery and evaluation. Our capabilities cover target protein ligand design, linker optimization, computational structure analysis, protein interaction modeling, binding affinity measurement, ternary complex assay support, cellular activity evaluation, and physicochemical property assessment. These services help researchers explore induced proximity strategies for cancer resistance models with greater efficiency and flexibility.

Target Protein and Ligand Design Support
- Ligand design for target protein helps identify and optimize target-binding ligands suitable for RIPTAC-inspired bifunctional molecule construction.
- Ligand for target protein products support early target engagement studies, molecular design exploration, and compound screening workflows.
- Target-focused design strategies can be adapted to resistant cancer models where differential protein expression is used as a selective recognition feature.
Linker and Bifunctional Molecule Optimization Support
- Linker design and optimization services support linker length, polarity, rigidity, flexibility, and exit vector optimization for induced proximity molecule design.
- Linker library resources enable rapid exploration of linker chemotypes for ternary complex formation and cellular activity studies.
- PROTAC linker products provide practical building blocks that can support bifunctional molecule synthesis and optimization.
Structure-Based and Computational Design Support
Binding, Complex Formation, and Cellular Evaluation Support
Frequently Asked Questions (FAQ)

Still have questions?
Contact Us
How do RIPTACs overcome cancer treatment resistance?
RIPTACs are designed to address resistance by shifting the therapeutic logic from simply blocking a cancer driver to inducing proximity between a tumor-enriched target protein and an essential effector protein. When both proteins are present in the same cancer cell, the RIPTAC molecule can promote a ternary complex that functionally disrupts the effector protein and drives selective cancer cell death. This mechanism may remain useful even when the tumor target is not the primary disease driver, which is why RIPTACs are attracting attention in resistant cancer research.
How are RIPTACs different from PROTACs?
PROTACs usually recruit an E3 ligase to degrade a target protein, while RIPTACs are designed to bring a tumor-enriched target protein together with an essential protein and impair the essential protein’s function through induced proximity. This makes RIPTACs conceptually closer to a “hold and kill” strategy rather than a degradation-only strategy. For drug discovery teams, the difference matters because RIPTAC design depends heavily on target-protein ligand selection, effector-ligand selection, linker architecture, ternary-complex formation, and cell-context selectivity.
What targets are suitable for RIPTAC discovery?
Suitable RIPTAC programs often start with a tumor-enriched intracellular protein that can help localize activity to cancer cells, even if that protein is not itself the core oncogenic driver. The effector side typically involves a protein whose functional inhibition can strongly affect cancer cell survival. BOC Sciences can support early discovery needs such as ligand sourcing, bifunctional molecule design, linker exploration, custom synthesis, analog library preparation, and structure-activity relationship support to help teams evaluate whether a proposed target-effector pairing is chemically and biologically tractable.
What assays support RIPTAC optimization programs?
RIPTAC optimization usually requires assays that connect chemistry with mechanism, including target engagement, ternary-complex formation, protein-protein proximity, pathway modulation, cell viability, selectivity profiling, and resistance-model testing. Because small changes in linker length, exit vector, rigidity, or ligand affinity can strongly affect ternary-complex behavior, iterative design-test cycles are important. BOC Sciences can assist with customized assay planning, compound-library synthesis, bioconjugation-related chemistry, cellular activity evaluation, and analytical support so discovery teams can compare candidates systematically before advancing the most promising chemical series.
Why partner with BOC Sciences for RIPTAC development?
RIPTAC discovery is a multidisciplinary workflow involving medicinal chemistry, linker engineering, ligand optimization, induced-proximity biology, and mechanism-focused testing. BOC Sciences can help drug developers reduce technical friction by supporting custom synthesis of heterobifunctional molecules, linker and warhead modification, focused analog generation, reference compound preparation, and in vitro evaluation strategies. This integrated support is especially useful for teams exploring novel RIPTAC concepts, validating target-effector combinations, or expanding from a proof-of-concept molecule into a broader optimization campaign.
Explore More
Discover More Research Products
Explore featured products that can expand your research options and accelerate your next discovery.
Expert Services to Move Your Project Forward
Access end-to-end service solutions that help bring efficiency, flexibility, and expertise to your research pipeline.
News
Technical Information
References
- Békés, Miklós, et al. "Regulated Induced Proximity Targeting Chimeras: A Heterobifunctional Small-Molecule Strategy for Selective Cancer Cell Killing." Cell Chemical Biology, 2024. https://www.cell.com/cell-chemical-biology/fulltext/S2451-9456(24)00307-6
- "RIPTACs for Precision Cancer Research: Target Protein and Essential Protein Proximity-Based Mechanisms." PubMed Central, National Center for Biotechnology Information, https://pmc.ncbi.nlm.nih.gov/articles/PMC12507177/
- "Regulated Induced Proximity Targeting Chimeras—RIPTACs—A Heterobifunctional Small Molecule Strategy for Selective Cancer Cell Killing." ScienceDirect, Elsevier, https://www.sciencedirect.com/science/article/pii/S2451945624003076

