As a leading service provider in drug discovery and research, BOC Sciences is fully capable and committed to providing one-stop proteolysis targeting molecular drug discovery based on chimeric (PROTAC®). With a comprehensive and advanced platform, we provide molecular docking services for protein-ligand to customers around the world to meet new drug discovery goals.
Introduction
Molecular docking is an effective method to study protein-ligand interaction and recognition. The near-natural conformation formed in the process of intermolecular interaction is a conformation with extremely low binding free energy. Searching the conformation with extremely low energy quickly and accurately is very important for protein-ligand molecular docking. Protein-ligand molecular docking conformational search methods mainly include fast exhaustive search and heuristic search. If the ligand is a biological macromolecule, such as protein, RNA or DNA, the interaction region can appear anywhere on the molecular surface, so it is often necessary to conduct a global search, which can use fast exhaustive search to traverse various locations, or heuristic algorithms for approximate global search. If the ligand is a small molecular compound, the molecular docking often has a certain binding pocket, which can limit the search range of the conformational space, and the heuristic algorithm is usually used to search the limited region.
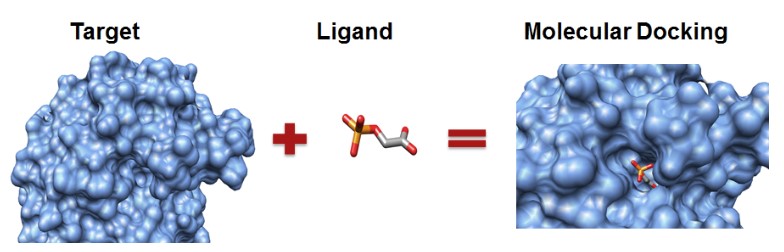
Our Services
- If there are missing atom or residue in the receptor or ligand protein, it needs to be filled.
- Only atoms or residues of standard amino acids are allowed in proteins, and atoms in non-standard residues are treated as carbon atoms with zero charge.
- In general, water molecules should be removed before calculation. However, if water is included, the oxygen atom is treated the same as the unknown atom (that is, as carbon with zero charge).
- If the input ligand or receptor has multiple conformations (for example, the structure from the NMR experiment), the first conformation will be used in the calculation. If conformations other than the first conformation should be used for calculation, all conformations other than the specified conformation should be deleted.
- Regardless of whether there is a hydrogen atom in the input molecule or not, the hydrogen atom will not be used in the calculation.
Our Advantages
- 100% Post Service Assistance
- Free Services Training
- Creativity and chemistry/biology problem solving
- Weekly progress reports
- Competitive pricing and fast turn-around time
- Molecular modeling and computational chemistry procedures with modern state-of-the- art software and algorithms
References:
- Morris, G. M., & Lim-Wilby, M. (2008). Molecular docking. In Molecular modeling of proteins (pp. 365-382). Humana Press.
- Drummond, M. L., & Williams, C. I. (2019). In silico modeling of Protac-mediated ternary complexes: validation and application. Journal of chemical information and modeling, 59(4), 1634-1644.
* PROTAC® is a registered trademark of Arvinas Operations, Inc., and is used under license.
