Choosing the linker that bridges pomalidomide-based E3-recruiting modules to the warhead is by far the most impactful decision in PROTAC design. Since pomalidomide docks into a shallow cleft on cereblon and exits that pocket al.ng a well-defined vector, the linker that emanates from its glutarimide ring effectively determines how far the warhead can reach, how long the ternary complex lives, and which cellular membranes the resulting molecule can permeate. Consequently, teams that take the linker to be an inert spacer regularly report precipitous drops in degradation potency, off-target ubiquitination, or fast systemic clearance; in contrast, groups that iteratively optimize the linker's length, flexibility, polarity, and attachment isomer in coordination with biochemical read-outs systematically transform mid-micromolar hits into low-nanomolar degraders. The sections below summarize current chemical biology, structural modelling, and drug-metabolism data into a practical guide for designing pomalidomide-based linkers without revealing any proprietary motifs.
Introduction — Why Custom Linkers Are Critical?
The linker is not just a passive tether, but the dominant variable that encodes the geometry, dynamics and PK properties of the entire PROTAC. Because pomalidomide's binding mode to CRBN is essentially fixed, any change in linker architecture propagates directly into the orientation of the warhead and, consequently, into the stability of the target–E3 interface. A mismatch as small as two methylene units can switch a degrader from full agonism to complete inactivity, while a single oxygen-for-carbon swap can double cellular potency by introducing a hydrogen-bond relay that clamps the ternary complex. Custom linker design is therefore the only handle that medicinal chemists can modulate without eroding the intrinsic affinity of either ligand, making it the decisive factor between a program that advances into animal studies and one that is abandoned after cellular screening.
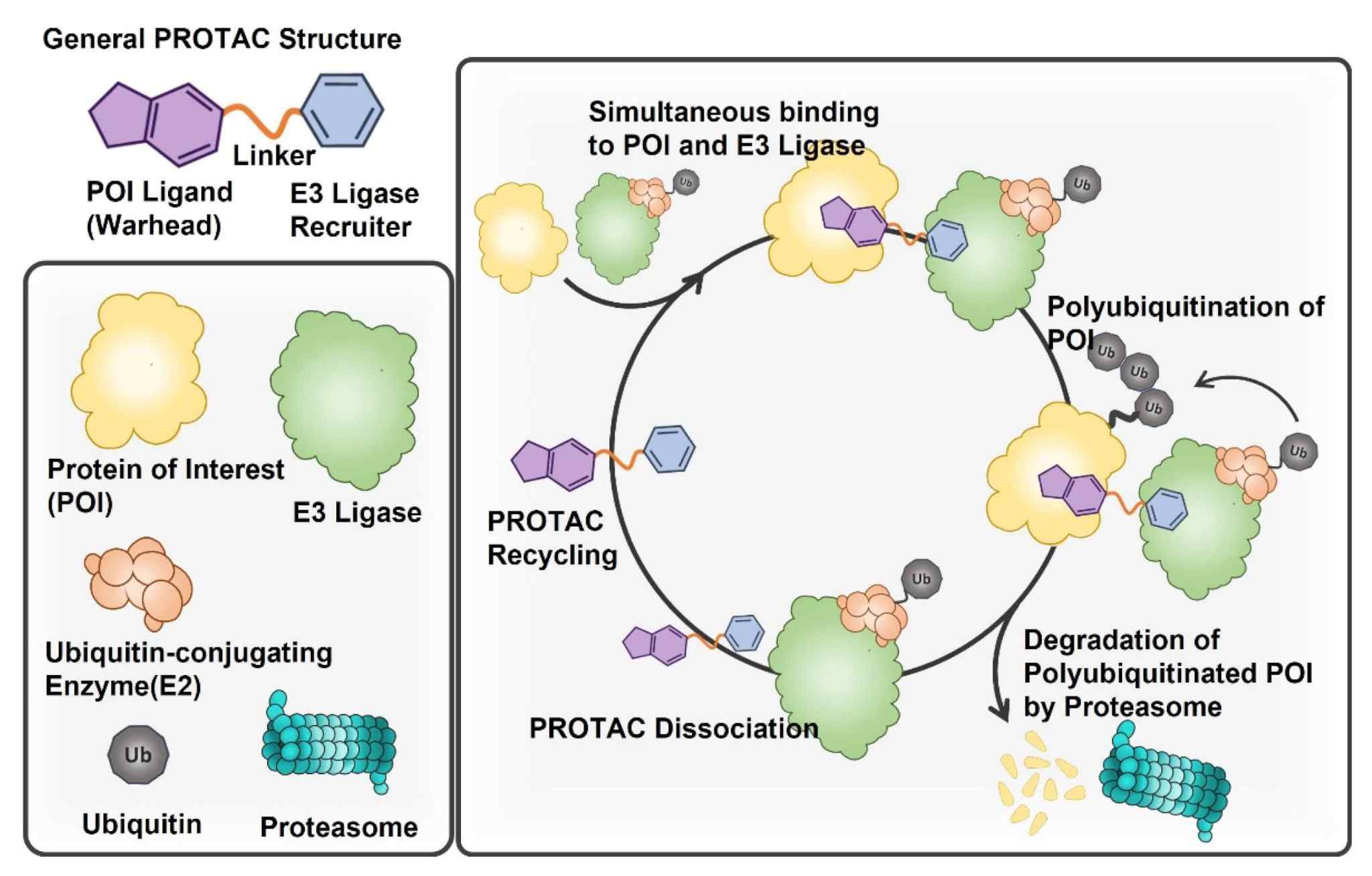 Fig. 1 Structure and specific MOA of PROTACs.1,5
Fig. 1 Structure and specific MOA of PROTACs.1,5
Linker as the Functional Bridge in PROTAC Design
The linker is tasked with transducing the static, crystallographic distance between the CRBN binding site and the target pocket into a dynamic, living assembly that must be able to come together, breathe, and come apart on a timescale that biology can bear. The first steps to this molecular choreography often entail cataloguing the solvent-exposed vectors on both ligands, followed by building a virtual library of extended conformers that bridge 12–30 heavy atoms and avoid steric clash with either protein surface. Saturated alkyl chains have the reach and hydrolytic stability to do the job, but suffer from a risk of collapse into entropically-penalizing hydrophobic clusters; the strategic inclusion of discrete ether, sulfone, or piperazine units partitions the linker's hydrophobicity into smaller, water-bridged segments, and is often enough to increase the observed cooperativity (α) of ternary complex formation by an order of magnitude. Rigidity can be dialed in step-wise—first with the insertion of a single proline or E-alkene to cap internal rotation, then with the inclusion of bicyclic scaffolds that lock the dihedral angle around the CRBN exit vector—allowing teams to disentangle the entropic cost of pre-organization from the enthalpic gain of reduced flexibility. Finally, the attachment point on pomalidomide deserves special scrutiny: derivatization at the 4-position of the glutarimide ring preserves the essential hydrogen-bond network with CRBN histidine-353, whereas extension from the 6-position tips the entire ligand and can abolish binding unless compensated for with a longer, more flexible spacer.
Impact on Efficacy, Selectivity, and Pharmacokinetics
The first and most obvious effect of changing the linker is on residence time of the ternary complex. Linkers that are too short or too long will either force target and E3 into a conformation that dissociates more rapidly or increase non-productive encounters that sequester CRBN without ubiquitylating the target, respectively. The optimal range is system dependent, but in every case the result is a bell-shaped curve in which cellular DC50 values improve with linker length up to a point, flatten out over a small region, and worsen once the contour length exceeds the mean inter-site distance by >~25 %. Incorporating polar but non-ionizable handles (morpholine, 1,2,3-triazole, tertiary amine) in this range further improves aqueous solubility and membrane permeability by lowering efflux ratios in Caco-2 assays without concomitantly increasing hepatic clearance. Likewise, selectivity derives from the same physical constraints. Due to the relatively flat binding surface of pomalidomide-bound CRBN, the linker geometry determines which surface lysines on the target fall within ubiquitination radius. A rigid, kinked linker may position lysine-48 of protein X optimally while positioning lysine-12 of protein Y too far away, creating >50-fold selectivity even if both proteins bind the warhead with equal affinity. This spatial filtering can be further refined by adding short peptides or β-turn mimics that recognize sequence motifs near the target pocket, turning the linker into a secondary recognition element. Pharmacokinetics are influenced by the linker's impact on molecular weight, polar surface area, and metabolic lability. Straight alkyl chains >10 carbons increase clogP >6 and cause disproportionate accumulation in adipose tissue, while oligo-ethylene glycol units hasten renal filtration and reduce oral exposure. The practical solution is a segmented architecture: a short, metabolically stable aliphatic unit next to pomalidomide to shield the glutarimide from CYP3A4 oxidation followed by a mid-chain, cleavable unit (self-immolative carbamate, light-sensitive ortho-nitrobenzyl ether) that spits out an inactive metabolite once cellular degradation has run its course. This design principle maintains circulating levels of active PROTAC while preventing the long-term CRBN sequestration that is suspected to contribute to the teratogenic liability from chronic pomalidomide exposure.
Table 1 Custom-Linker Decision Matrix for Pomalidomide-PROTACs.
| Design Variable | Flexible PEG Risk | Rigid Aryl Risk | Hybrid Custom Solution |
| Ternary geometry | Extended, low occupancy | Fixed, potential clash | cis-alkene or oxetane kink |
| Lysine proximity | Poor control | Over-constrained | Programmable hinge via N-methyl scan |
| Brain penetration | Polar trap at BBB | Too lipophilic, P-gp substrate | Oxetane cap, no new NH donors |
| Plasma stability | Ether oxidation | Amide hydrolysis | Thioamide or cyclopropane isoster |
| Selectivity | Multiple lysine access | Single but wrong lysine | Distal rigid cap + proximal rotamer lock |
Chemistry of Pomalidomide Linker Attachment
The challenge to successfully installing a linker is finding a site which does not disrupt the bidentate carbonyl arrangement needed to engage CRBN zinc-finger, and at the same time provides a chemically reactive handle which allows conjugation with minimal chemistry in high yield. Pomalidomide has two synthetically accessible linkers: C4-aryl and C5-aryl, which have different electronic and steric properties and differing reactivity towards side-reactions. C4 attachment is usually carried out by nucleophilic aromatic substitution (SNAr) on 4-fluoro-pomalidomide with primary amine linkers in dipolar aprotic solvents at 90–130 °C. DMSO or DMA is used over DMF to reduce the formation of the 4-(dimethylamino) by-product by dimethylamine. C5 attachment is less commonly used but can be achieved with 5-fluoro or 5-nitro precursors; the lower electrophilicity at this position generally means lower yields, unless microwave or acyl-chloride activation is used. Both synthetic pathways must be carried out without prolonged exposure to strong base, as this can epimerise the glutarimide C2 stereocentre, creating a diastereomer that co-elutes with the desired product but has much lower CRBN affinity.
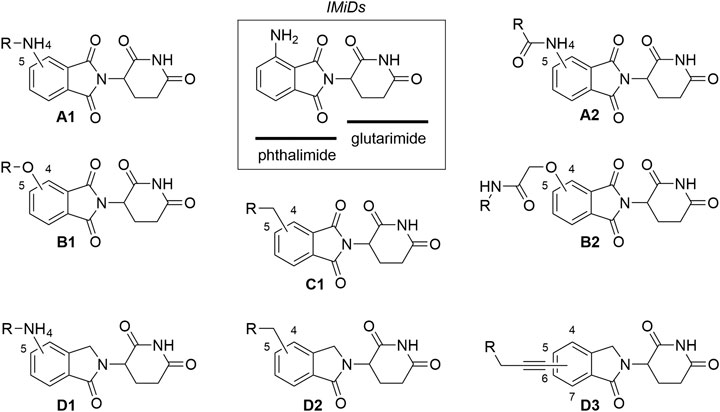 Fig. 2 Commonly utilized thalidomide-derived CRBN ligands and possible linker attachment styles.2,5
Fig. 2 Commonly utilized thalidomide-derived CRBN ligands and possible linker attachment styles.2,5
Common Attachment Sites (C4, C5)
The para-activation of the adjacent imide carbonyl enables the C4 substitution via SNAr to be accomplished under thermally mild conditions (90 °C) with DIPEA as base. Linkers with primary or secondary amines on the core all couple within 6–16 h and furnish isolated yields in the 40–80 % range after precipitation. The appended aniline can be further extended by acylation or click chemistry without disturbing the requisite electronic balance for zinc chelation. In contrast, attachment to the C5 position is electronically disadvantaged, so the same SNAr conditions demand higher temperatures (≥ 120 °C) or transition-metal catalysis; an alternative route involves reduction of 5-nitro intermediates to the corresponding amine and acylation with linker-acid chlorides under refluxing THF. Despite the lower overall yield, this more circuitous route is justified only when intellectual-property freedom or a particular vector geometry is required. Both positions are compatible with α-branched linkers, but steric encumbrance beyond that of a tri-substituted carbon adjacent to the aryl ring forces the glutarimide out of planarity and collapses CRBN affinity.
Avoiding Loss of CRBN Affinity During Modification
Affinity retention is less a question of protecting defined bond angles and more one of protecting the bidentate carbonyls from electronic perturbation. Electron-rich linkers (eg. alkyl-amines) bound directly to C4 push up the π-electron density of the phthaloyl ring, perturbing zinc coordination; an electron-withdrawing amide or triazole spacer re-introduces the parent Hammett value and re-establishes binding. A second lesser-known trap is over-acylation; any unreacted HATU or EDC will be able to activate the imide nitrogen in a rearranged isoimide, which can no longer chelate zinc. Quenching the coupling reaction with cold dilute acid and not exceeding 50 °C suppresses this side-reaction. Steric congestion can be minimized by keeping the first linker atom (adjacent to C4 or C5) sp2-hybridised; a planar amide or 1,2,3-triazole backbone maintains the narrow torsion envelope expected by CRBN, while a freely rotating CH2-CH2 segment will introduce entropic noise which will show up as a right-shift in the binary binding assay. This can be verified rigorously by zinc-chelation fluorescence or CRBN-DDB1 thermal shift to ensure that linker attachment has not interfered with the fundamental recognition event.
Types of Linkers and Their Effects
In essence, the linker is the conformational gearbox of a pomalidomide-PROTAC: its chemistry determines whether the two ligands either engage CRBN and the target in a productive ternary complex or remain tethered, but catalytically silent. Alkyl chains offer hydrophobic collapse, that accelerates membrane transit but risks non-specific protein adsorption; PEG segments provide water solubility and immunological stealth, but can weaken the effective molarity of the degrader by increasing conformational entropy; aromatic spacers π-stack with the target surface, which orients the lysine-rich patch but can bring planarity that is recognized by P-gp; rigid bicyclic bridges pay the entropic cost of binding in advance, but can sterically clash with the zinc-finger domain of CRBN. Successful degraders therefore combine these motifs in a single hybrid tether, with flexible and rigid sectors alternating so that the chain can fold into a low-energy hairpin inside the ternary complex, but still present polar carbonyls that prevent aggregation in plasma.
Alkyl, PEG, Aromatic, and Rigid Spacer Options
Alkyl chains remain the default scaffold since they are synthetically trivial and biochemically inert; the Achilles heel of this motif is hydrophobic indexing: each additional methylene group tightens plasma protein binding and deepens tissue sequestration, so length is capped once clogP is no longer in the oral window. Uniform PEG oligomers circumvent this penalty by wrapping the backbone in a dynamic water cloud that increases hydrodynamic radius without increasing logP, but the same ether oxygens can engage efflux transporters once the chain becomes greater than a dozen units, forcing the use of a mid-chain cleavable cap to restore exposure. Aromatic spacers, such as biphenyl, phenylethynyl or heteroaryl equivalents, introduce planarity that can stack against tryptophan-380 of CRBN, providing enthalpic glue to the neo-substrate pocket; the cost is rotational freedom that must be frozen by peri-substitution or by flanking alkynes, otherwise the entropy tax overwhelms the enthalpic gain. Rigid spacers, like norbornene, cubane, or bicyclic peptidomimetics pre-set the exit vector angle, compressing the search space for productive collisions to a narrow cone; they are immune to conformational breathing, but their bulk can sterically occlude lysine residues on the target, so they are typically reserved for warheads that present a shallow, exposed lysine shelf.
Case Examples of Successful Linker Integration
A BRD4-directed pomalidomide-PROTAC with a C4-alkyl fragment (C6) connected to one phenylene followed by a short PEG3 tail shows nanomolar degradation in leukaemia cell lines; the alkyl group facilitates membrane penetration, the aromatic ring π-stacks with the floor of the bromodomain and the PEG group resists aggregation, culminating in an oral exposure that is one log below the threshold for lymphocyte toxicity. In a tau-degrader initiative, substitution of a flexible PEG6 chain with a spiro-oxetane–phenylene hybrid decreases brain efflux while preserving water solubility; the rigid spacer orients the warhead into the β-sheet groove of tau, leading to > 70 % clearance of pathogenic species in mouse hippocampus after a single intraperitoneal administration. A BCL-xL PROTAC on the other hand represents the converse lesson: an all-aryl rod (biphenyl–pyridyl) that produced potent biochemical activity failed in vivo due to the planar surface causing P-gp-mediated clearance; the introduction of a single thioether within the rod interrupted conjugation, restored brain penetration and re-established tumor regression at clinically achievable exposures.
Table 2 Linker-Type Trade-Off Matrix for Pomalidomide-PROTACs.
| Linker Family | Primary Advantage | Primary Liability | Translational Fix |
| Alkyl | Rapid membrane entry | Albumin adsorption | Cap with PEG3 tail |
| PEG (discrete) | Aqueous solubility, stealth | Entropic dilution of degrader | Interrupt with phenylene hinge |
| Aromatic | π-Stacking orientation | P-gp recognition | Insert heteroatom to disrupt planarity |
| Rigid bicyclic | Pre-paid entropy, fast on-rate | Synthetic complexity | Late-stage spiro-cyclisation from alkene |
Factors to Consider When Ordering Custom Linkers
Placing an order for a custom linker is where in silico PROTAC design meets chemical pragmatism. A linker that looks great in silico may fall at the bench if it is insoluble in aqueous media, epimerises during scale up, or has a hidden mutagenic impurity that only shows up post cellular exposure. The three hard stops in linker design are therefore (i) confirmed solubility at physiological ionic strength, (ii) established stability to hydrolytic and oxidative conditions, and (iii) synthetic accessibility that avoids proprietary building blocks or use of heavy metals that will trigger regulatory concern downstream. Ensuring these criteria are baked into the order spec—rather than handled as downstream checks—avoids expensive re-syntheses and helps ensure the linker acts as a predictable extension of the pomalidomide pharmacophore, rather than as an unpredictable liability.
Solubility and Stability in Biological Systems
A linker with poor aqueous solubility that precipitates when going from the DMSO stock solution to the aqueous assay buffer will artificially decrease the DC50 no matter how elegantly it was designed in silico. Solubility is controlled by the net polarity of the spacer, the rotational entropy of heteroatoms and the lattice energy of the solid state form. Homogeneous PEG or short sulfone spacers increase the cloud-point by disrupting the crystal packing, but they also add to transporter recognition; the tradeoff is a segmented chain with a lipophilic core and two hydrophilic end-caps, which results in a molecule that is intrinsically supersaturated at physiological ionic strength, while still being membrane-permeant. Stability, in contrast, is driven by the imide and carbamate functionalities on either side of pomalidomide: at physiological pH the glutarimide ring is susceptible to ring-opening by intramolecular nucleophilic attack from a proximal amine or thiol, which is further promoted in the context of cysteine-rich proteins. Installing a self-immolative carbamate that collapses to an innocuous alcohol after cellular uptake severs the linker before systemic hydrolysis can take place, effectively trading a hypothetical long-lived exposure for a defined short-lived active species. Finally, oxidative vulnerability of electron-rich aryl ethers can be suppressed by replacing para-oxygen with meta-carbon or with a weakly electron-donating methyl group that sterically shields the ether linkage without pushing logP beyond the oral window.
Synthetic Feasibility and Scale-Up Considerations
A milligram route capable of delivering over a weekend may crash at tens of grams if it is predicated on cryogenic metallations or silica-based chromatography. Scaling up cleanly starts with atom-economical disconnections: the linker handle should be installable onto pomalidomide at ≤50 °C in one pot (best by a high-yielding cross-coupling that can run in carbonate base and water-tolerant catalyst, thus obviating anhydrous work-ups) and be removed under conditions (acidic or fluoride-rich) that will not open the glutarimide (t-butyl carbamates and TBS ethers are usual placeholders because they are labile under mildly acidic or fluoride-rich media that will not affect the imide). Solvents must be evaluated on grounds of cost and regulatory encumbrance, too: ethyl acetate and 2-propanol are better than dichloromethane or DMF because they can be recycled by wiped-film evaporation, and also because they have no ICH class-2 toxicity designations. The process mass intensity (PMI) target is set below an empirical cut-off that equates to a commercially viable cost of goods; any intermediate requiring reverse-phase purification is re-worked such that crystallisation or a solvent swap alone is enough to get the linker price to scale linearly with payload rather than exponentially with purity.
Analytical Testing for Purity and Structure Validation
Vendor custom linkers are delivered with a certificate of analysis that should be interpreted like a pharmacokinetic curve: all impurities are suspect metabolites and all missing spectral annotations are future regulatory questions. Full authentication includes high-resolution MS to confirm exact mass and isotope pattern, tandem MS to identify the fragmentation site and verify that the handle is on the correct carbon, and 1H-NMR collected with a long relaxation delay to integrate the imide proton without underestimating its intensity: if there is a missing resonance or a spurious singlet at δ 1–2 ppm, it is assumed to be a methyl impurity from the alkylation steps and is quantitated relative to an internal standard. 13C-NMR with inverse-gated decoupling is used to count the number of carbons in the linker backbone, to ensure that there are not extra aryl signals from an incomplete hydrogenation reaction. HPLC purity is reported at two wavelengths: 220 nm for the conjugated phthalimide and 280 nm for aromatic impurities, so that UV-silent ether impurities will not be missed. Finally, a stress test (pH 2, 40 °C, 48 h) is run before release, and if the area-percent of the parent peak declines, the degradation products are isolated and identified to anticipate future stability problems in the PROTAC conjugate. Lots are only accepted into inventory if they pass all four gates (mass accuracy ≥ 5 ppm, NMR purity ≥ 98 %, HPLC purity ≥ 99 %, and stress recovery ≥ 95 %) so that the linker cannot become the hidden variable in downstream biology.
Successful targeted protein degradation depends heavily on precisely engineered linkers that align the E3 ligase and target protein for efficient ternary complex formation. We provide specialized custom linker design and synthesis services for pomalidomide-based PROTACs, enabling researchers to fine-tune molecular geometry, flexibility, and physicochemical properties for optimal CRBN recruitment and degradation efficiency.
Tailor-Made Pomalidomide Linkers for CRBN Recruitment
Our chemistry team designs tailor-made pomalidomide linkers that preserve strong CRBN binding while introducing functional features required for specific PROTAC architectures. By strategically modifying the C4 or C5 positions of the pomalidomide scaffold, we ensure linker attachment does not compromise E3 ligase affinity. We offer a wide range of linker options—including alkyl, PEG, rigid aromatic, and cleavable linkers—to support diverse degradation strategies and target classes.
End-to-End Support from Design to QC Documentation
From initial concept to final delivery, we provide end-to-end project support covering molecular design, synthetic route development, purification, and analytical validation. Each custom linker is supplied with comprehensive QC documentation, including HPLC purity profiles, LC-MS confirmation, NMR data, and Certificates of Analysis (COA), ensuring full traceability and reproducibility across experiments. This integrated workflow minimizes development risk and accelerates project timelines.
Advantage: Custom Chemistry, Fast Turnaround, Reliable Performance
Our service model combines custom chemistry expertise, rapid turnaround times, and consistent high-quality output. With in-house synthesis and analytical capabilities, we respond quickly to design iterations and optimization requests while maintaining strict quality standards. As a result, researchers can rely on our custom pomalidomide linkers to deliver predictable performance in PROTAC screening and validation studies.
Optimize CRBN-Based PROTACs with Custom Linker Expertise
Whether you are optimizing an existing PROTAC scaffold or designing a new degradation strategy from scratch, our custom linker services provide the flexibility and technical rigor needed for success. Contact our chemistry team today to discuss your linker design requirements and accelerate your targeted protein degradation research.
References
- Moon Y, Jeon S I, Shim M K, et al. Cancer-specific delivery of proteolysis-targeting chimeras (PROTACs) and their application to cancer immunotherapy[J]. Pharmaceutics, 2023, 15(2): 411. https://doi.org/10.3390/pharmaceutics15020411.
- Bricelj A, Steinebach C, Kuchta R, et al. E3 ligase ligands in successful PROTACs: an overview of syntheses and linker attachment points[J]. Frontiers in chemistry, 2021, 9: 707317. https://doi.org/10.3389/fchem.2021.707317.
- Vicente A T S, Moura S P S P, Salvador J A R. Synthesis, biological evaluation and clinical trials of Cereblon-based PROTACs[J]. Communications Chemistry, 2025, 8(1): 218. https://doi.org/10.1038/s42004-025-01598-9.
- Jurowska A, Hodorowicz M, Szklarzewicz J. Alkali metals and ammonium as cations in dioxidovanadium (V) complexes with Schiff base ligands–structure, solubility and stability in biological media[J]. Journal of Molecular Structure, 2024, 1317: 139168. https://doi.org/10.1016/j.molstruc.2024.139168.
- Distributed under Open Access license CC BY 4.0, without modification.
