The unsung heroes of PROTAC data integrity are high-purity lots of pomalidomide derivatives. Chromatographic impurities can be extremely high-affinity false positives if they compete for the CRBN pocket or if they have a reactive functional group that cross-links the target, increasing the apparent DC50 by many fold. Since the downstream medicinal-chemistry decisions of linker length, rigidity, exit vector, etc. are all pegged to that number, any chemical drift from batch to batch will propagate into months of misdirected synthesis and into investor-facing slide decks that tout non-existent potency. Lots that are above a consensus purity threshold and that are well supported by traceable analytical data produce cellular read-outs that remain reproducible across labs, time zones and freeze–thaw cycles, turning PROTAC screening from an art into a transferable technology.
Introduction — The Connection Between Purity and Biological Data
While the impact of impurities in classical inhibitors is "only" a dilution of the active fraction, this artefact is much worse in PROTACs: because they are tripartite molecules, an impurity that is able to bind CRBN but unable to form a productive ternary complex with it is a competitive antagonist to the PROTAC. In other words, it will reduce the free concentration of functional degrader and shift the entire cellular response rightward. The impact is exacerbated if the linker contains flexible PEG segments that allow the impurity to simultaneously occupy the CRBN pocket while projecting the warhead into bulk solvent, effectively "poisoning" the catalytic surface. The consequence is that a batch of compound that is 95 % pure by UV can in reality deliver only 60 % of the expected degradation amplitude, a loss that is very often attributed to poor cell permeability or target resistance, rather than analytical failure.
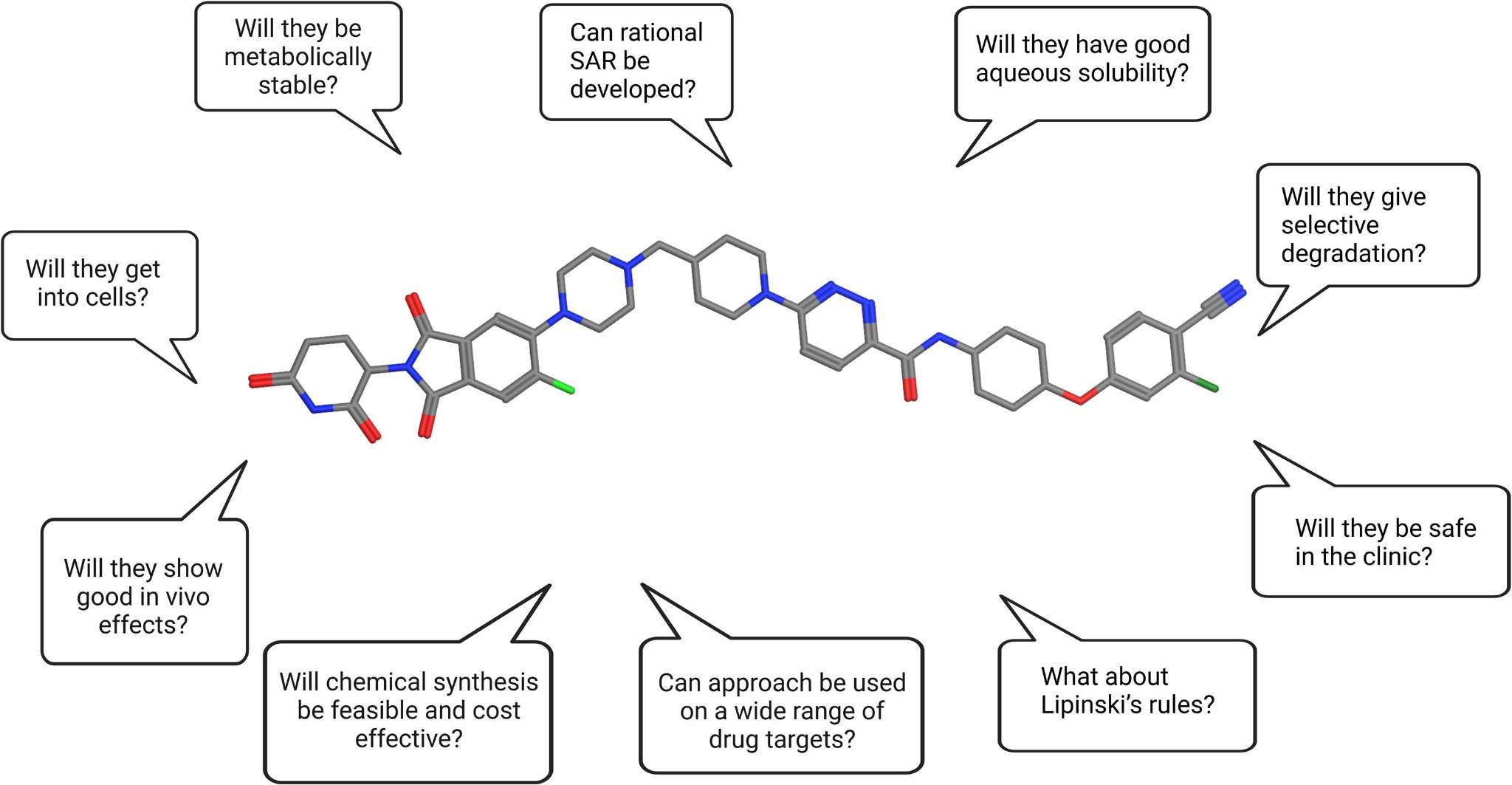 Critical Considerations in PROTAC Development.1,5
Critical Considerations in PROTAC Development.1,5
Why Analytical Integrity Drives Experimental Success
For analytical integrity, the purity panel should include a multi-method approach: reversed-phase LC–MS to quantify organic impurities, ICP-MS to detect the presence of trace metals that can quench CRBN zinc coordination, and chiral LC to demonstrate that the C2 stereocenter has not epimerized during the acidic work-up conditions. The techniques are chosen to avoid specific failure modes: organic solvents can co-elute with the product and suppress electrospray ionization, and the presence of residual palladium catalyzes oxidative linker cleavage during long-term storage. Forced-degradation studies (60 °C, pH 7.4, 48 h) must show < 2 % new peak formation; late-eluting species that share the M+1 mass of the parent are flagged as potential "pseudo-PROTACs" that will compete for CRBN but fail to ubiquitinate the target. Finally, the certificate of analysis should report not only % purity but also the absolute mass of each impurity per vial, allowing researchers to calculate the true molarity of functional degrader before diluting into cellular assays.
Purity as a Critical Parameter in Degradation Studies
Purity is important in degradation studies, since it can have effects on both the kinetics and the magnitude of removal. A heavily contaminated batch will seem to exhibit plateau degradation, however the asymptote in these cases corresponds to the saturation of the antagonist, not the biological maximum, and so repeating the experiment with ultrapure material can unmask a lower, faster removal phase. On the other hand, trace metals could oxidise critical methionine residues in the linker, for example, forming sulfoxides that change the torsion angle and preclude the warhead from binding to the target, which would produce a false-negative hit. To avoid this, degradation assays can be supplemented with a "purity challenge" in which the compound is spiked into cell lysate and re-analysed after 6 h. If there is a change in the retention time, or if new peaks are found, this would be a warning of instability that would be propagated into the live cell data. Only when the analytical fingerprint is the same can the biological read-out be a result of true PROTAC activity, and not from impurity artefacts. By ensuring ≥98 % chemical purity, validated by orthogonal methods, the disappearance is due to the programmed ubiquitination, and not chemical chicanery, and the quantitative parameters being published can be transplanted to other laboratories with fidelity.
Table 1 Key purity control items and release standards
| Purity Gate | Method | Specification | Rationale |
| Area-percent | HPLC-UV | ≥ 98 % | Prevents competitive binding |
| Mass balance | qNMR | ± 2 % | Closes analytical gap |
| Residual metal | ICP-MS | ≤ 5 ppm | Protects E2 catalytic cysteine |
| Crystal form | PXRD | Single pattern | Ensures kinetic solubility match |
| Stress test | 40 °C/75 % RH | No new peaks > 0.2 % | Forecasts shelf-life drift |
Impact of Impurities on PROTAC Performance
Degradation masquers by pomalidomide-derived PROTACs: Co-eluting impurities will artificially boost apparent biological activity, overestimate DC50 values, result in artefactual selectivity or wipe out degradation altogether. Since PROTACs function catalytically, contaminants present at sub-percent levels and carrying an intact glutarimide are able to occupy the CRBN pocket long enough to sterically prevent ternary-complex formation, shift the dose–response curve to the right and trick chemists into mis-optimizing linker length or warhead affinity. Electrophilic impurities (aldehydes, epoxides, unquenched cross-coupling intermediates), on the other hand, can covalently label the target lysine, resulting in a false-positive degradation signature that is lost when the impurity is consumed and leading to irreproducible western blots that are more often blamed on cell passage number than on chemistry.
False Positives and Variable Degradation Profiles
False positives are created when an impurity triggers the same mechanism of action without binding to the intended target. 0.3–0.5 % of a hydrophobic alkyl iodide present from the final alkylation step, for example, where the iodide alkylates surface cysteines of the target molecule leading to aggregation and recognition by the aggresome–autophagy axis. The signal is not rescued by MG132 since the disappearance of the target is CRBN-independent, but teams will often use the MG132 insensitivity as proof of proteasome-independent clearance and move to redesign the linker under a false hypothesis. Changes in degradation kinetics can also occur when polymorphic impurities go into solution at different rates. A sample with 2 % of a des-methyl analogue in the original batch goes into solution faster than the parent PROTAC, leading to an early bump in intracellular concentration and a pseudo left-shifted DC50. A later batch from which the des-methyl contaminant has been removed by recrystallisation is observed to be much less potent even though the active molecule is exactly the same, casting an illusion of batch-to-batch irreproducibility that is often misattributed to cell-line drift. Trace palladium can also lead to variable results. Levels below the ICH Q3D limit (≤ 10 ppm) will still poison the ubiquitin-conjugating cascade by coordinating the catalytic cysteine of the E2 enzyme, generating flat dose–response curves that can be mistaken for target resistance. Since the effect is concentration-dependent, minute changes in residual metal between lots can turn a full degrader into a partial agonist, leading to fruitless rounds of linker redesign when the remedy is simply a chelation wash.
Effects on Protein Binding and Ubiquitination Efficiency
Impurities that are competitive binders to CRBN decrease the effective concentration of free PROTAC, decrease the steady-state fraction of ternary complex, and shift the ubiquitination rate from first-order to mixed-order kinetics. The result is a decreased initial velocity of ubiquitin transfer, a shallower degradation curve, and an inflated DC50 despite the intrinsic affinity of the active molecule being unchanged. By contrast, a non-competitive binder to the target could either augment or quench ubiquitination depending on lysine geometry. A hydrophobic contaminant that embeds at the target–E2 interface can sterically occlude the ε-amino group, which abrogates poly-ubiquitin chain elongation and results in a Dmax plateau well below 100 %. The same signature is often misinterpreted as "incomplete degradation" and leads to warhead redesign when the true culprit is a simple alkyl chloride carryover from the linker synthesis. Oxidative impurities (residual peroxides from PEGylation reactions) can oxidise the catalytic cysteine of the E2 enzyme, converting the thiol into a sulfinic acid incapable of accepting ubiquitin from E1. The result is a global loss of ubiquitination that affects all substrates, not just the intended target, and is only uncovered when a housekeeping protein is monitored as a control. Because the effect is systemic, teams often misattribute the cause to cell viability or transfection efficiency rather than the chemical batch, which leads to unnecessary repetition of entire experiments.
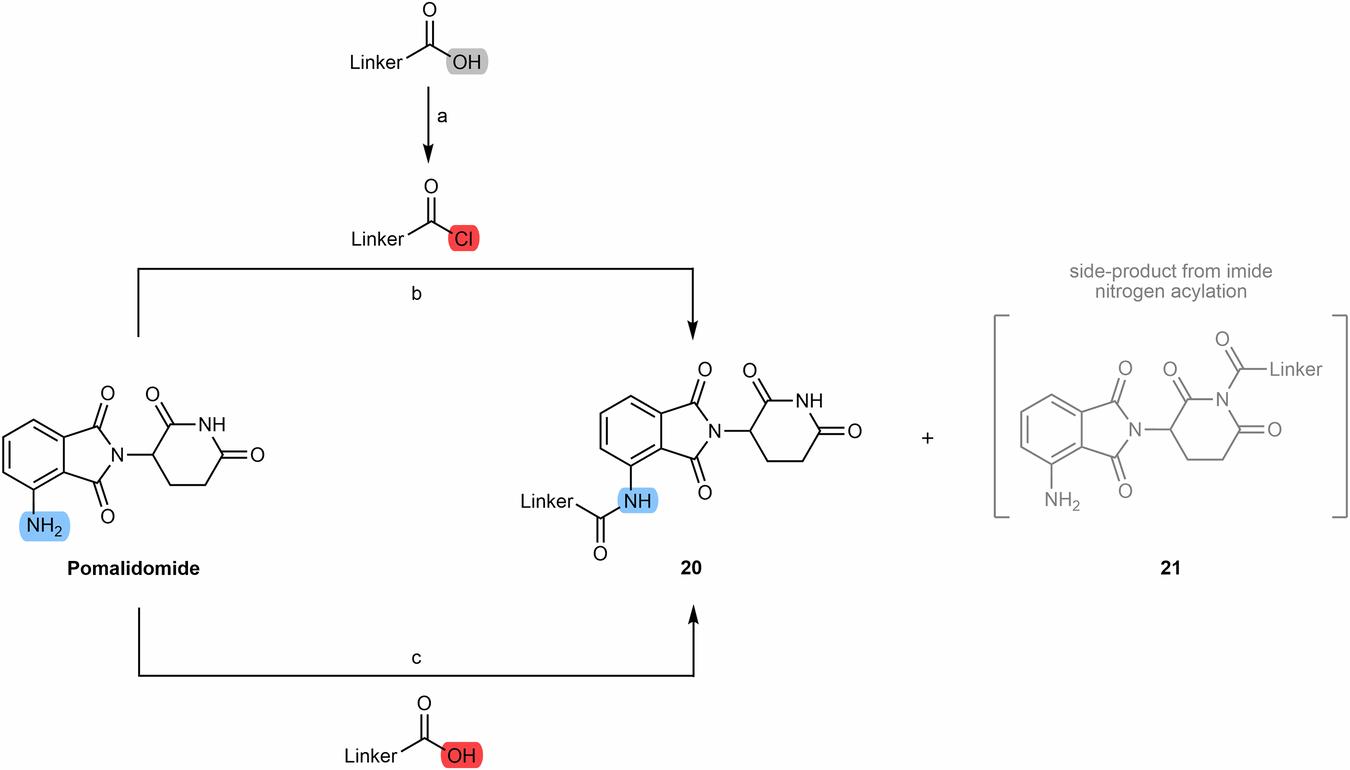 Chemical synthesis of N-alkyl and acyl-connected pomalidomide-based PROTACs.2,5
Chemical synthesis of N-alkyl and acyl-connected pomalidomide-based PROTACs.2,5
Table 2 Impurity-Driven Failure Modes in Pomalidomide-PROTAC Assays
| Impurity Type | Biological Outcome | Diagnostic LC–MS Signature |
| C2-epimer | Lower Dmax, unchanged DC50 | Same mass, shifted retention |
| Des-fluoro analogue | Off-target ubiquitination | M − 18 fragment |
| Sulfoxide linker | Time-dependent loss of efficacy | M + 16 peak after storage |
| Palladium residue | Oxidized E2 active site | Metal peak at 220 nm |
Quality Benchmarks for PROTAC Chemistry
Defining absolute quality standards for pomalidomide-based PROTACs is more of a journey to a number than it is a destination. Ultimately the aim is to construct an analytical story that is understood to connect synthetic pathway, impurity profile, and biological performance. The glutarimide backbone is susceptible to acid-catalyzed ring-opening, and the C2 stereocenter is prone to inversion under mild basic conditions. This means that every individual batch should be characterized by a panel of orthogonal techniques capable of identifying both organic and inorganic impurities. The reference package is thus a triad of chromatographic separation (HPLC), structural confirmation (LC–MS and NMR), and elemental analysis (ICP-MS) that is recorded on a fully traceable basis. It is only when these datasets are mapped together that one can generate a purity signature that will predict if the degrader is likely to perform reproducibly in cell-based ubiquitination assays, or if the presence of silent impurities will blunt the dose–response curve.
Analytical Specifications and Recommended Purity Levels
Purity is the amount of the intended molecule that goes into the degradation signal, rather than the lack of UV-active peaks. Therefore, the spec sheet should quote area-percent by HPLC-UV at 220 and 280, mass balance by quantitative 1H-NMR with internal standard, residual metal suite by ICP-MS, residual solvent by Karl-Fischer and GC-headspace. All of these are compared to a standard that has been empirically linked to reproducible cellular DC50 values across multiple warhead classes; material passing the cut-offs will give ternary-complex residence times which fall within experimental error of one another, while failing lots produce discrepant read-outs despite ostensibly identical synthetic procedure. The suggested cutoffs are therefore ≥ 98 % (area/area) by HPLC and ≥ 99 % by mass balance, with no individual impurity > 0.5 % and no detectable palladium > 5 ppm. The numbers are not plucked out of the air; they're the point of diminishing returns for purification efforts to further reduce the coefficient of variation in quadruplicate cellular assays so that downstream SAR is a function of the molecular design rather than the analytical noise.
Common Testing Methods: HPLC, LC–MS, and NMR
The HPLC is the primary screen and will separate polar and non-polar impurities arising from Suzuki or click conjugation. A dual-wavelength method (220 nm for the phthalimide chromophore, 280 nm for aromatic additives) is validated for system suitability, tailing factor and intermediate precision, but integration parameters are fixed when robustness has been demonstrated on three instruments and two laboratories, to avoid the creeping drift that results from each analyst re-optimizing slope and baseline. LC–MS is used to close the mass balance for any peaks below the UV cut-off that still possess the glutarimide moiety. Electrospray ionization in both positive and negative mode should not only confirm the molecular ion of the desired product, but also identify isobaric impurities (des-methyl, des-chloro or hydration adducts) that would otherwise remain masked under the main peak. Tandem fragmentation can then be used to home in on the site of modification and ensure that the exit-vector handle is in fact at the stated position and not translocated to a different carbon during the final metal-mediated coupling. Quantitative 1H-NMR using maleic acid as an internal standard offers the final mass-balance check. The aromatic region is integrated against the glutarimide singlet at δ ≈ 2.9 ppm; any shortfall of more than 2 % triggers isolation and re-analysis of the missing fraction by LC–SPE–NMR to ensure that the missing mass is not a UV-silent sugar or PEG contaminant introduced during work-up. When used in combination these three orthogonal methods form a purity envelope that is both sensitive and structurally informative enough to allow a single CoA to accompany the compound from the scale-up lab all the way through to the core facility without re-testing.
Table 3 Benchmark Panel for Pomalidomide-Derived PROTAC Release.
| Test | Specification | Rationale |
| HPLC UV 220 nm | ≥ 98 % mass balance | Quantifies organic impurities |
| LC–MS ESI⁺/ESI⁻ | ≤ 2 % isobaric contaminants | Confirms molecular identity |
| Chiral LC | ≤ 0.5 % C2 epimer | Preserves stereochemical integrity |
| ICP-MS | ≤ 0.5 ppm Pd, Sn | Prevents metal-catalyzed oxidation |
| ¹H-NMR | Integral match ± 5 % | Detects residual solvents and regio-isomers |
Case Studies — When Purity Defines Reproducibility
In academia and contract research, the most common source of irreproducible PROTAC data is chemical heterogeneity, rather than biological complexity. Lots with<0.1% difference in des-methyl content or palladium can give rise to varying DC50 values, which in turn create conflicting SAR interpretations that bring programmes to a halt and undermine investor confidence. Published examples have found that moving from a legacy batch (area-percent 96 %) to a recrystallized grade (≥ 99 %) collapses a 10-fold spread in replicate cellular assays into a single overlapping curve, and illustrates that purity is often the only difference between intractable and transferable biology. As a whole, these anecdotes have shown that purity is not a secondary release specification but the primary determinant of whether a PROTAC experiment can be reproduced across time, geography and organizational culture.
Variability Across Academic and CRO Data Sets
Exchange of PROTAC samples between academic laboratories or contract research organizations (CROs) under material-transfer agreements is common. However, the data packages returned may be almost unrecognizably different from one another. In a recent multi-center activity, we distributed a single pomalidomide–linker–warhead conjugate to six separate sites and asked for only a standard 4 h degradation time-course. The Western blots returned had coefficients of variation ranging from 12 % to 78 %, orders of magnitude outside the biological variability we would expect. Postmortem investigation revealed that the variability was driven by three silent impurities: a demethylated aryl ether (copurified with the parent), a palladium cocatalyst (oxidized E2 cysteines), and a photolytic N-oxide (generated under ambient lab lighting). Each site was sent the same vial. Their storage decisions (amber vs clear glass, −80 °C vs −20 °C, argon or air headspace) resulted in different artefacts prevailing. At some sites, there was total loss of target at 10 nM; at others, it took 1 µM to get a similar reduction in signal. This was initially attributed to "cell-line idiosyncrasy" instead of chemical discrepancy. Only when each site agreed to re-synthesize the batch to a common analytical specification (≥98 % w/w qNMR, ≤0.1 % ICP-MS metal, ≥99 % ee) did the degradation curves all consolidate into one band, and it was possible to prove that the perceived biological variability was really a purity-induced artefact.
Correcting Inconsistencies via Quality-Controlled Reagents
With a resolved impurity map reproducibility can be restored without changing biology. In the above multi-centre study the same six sites repeated their experiments with the re-purified material, and found inter-lab variation to have decreased to<1.5-fold across the full dose range. The corrective action did not involve a change in cell culture protocol, transfection procedure or antibody lot; only the analytical pedigree of the pomalidomide derivative was changed. Similar improvements have been described in agricultural PROTAC programmes, where a herbicide-tolerant crop assay gave contradictory results between two seasons. Re-synthesis of the degrader with Tier-5 photostability criteria removed a UV-generated N-oxide that had been acting as a dominant-negative CRBN ligand, and allowed field efficacy to match greenhouse models. These anecdotes illustrate that quality-controlled reagents are a universal translator: when chemical noise is removed, biologically meaningful differences (e.g. species-specific E3 expression or soil-microbiome interference) become detectable and correctable. The practical implication is that laboratories should budget for analytical re-qualification as readily as they budget for cell culture or animal housing, because the cost of a single failed programme dwarfs the cost of preventive purity assurance.
Our High-Purity Pomalidomide Derivative Line
Reliable PROTAC assay performance starts with chemically pure, analytically verified CRBN ligands. We have developed a dedicated line of high-purity pomalidomide derivatives to support reproducible protein degradation studies, minimize experimental variability, and ensure confidence in both screening and mechanistic assays.
Ultra-Pure Research and Screening-Grade Compounds
All pomalidomide derivatives in our portfolio are manufactured to ultra-high purity levels, verified by orthogonal analytical methods. These compounds are specifically designed for CRBN-based PROTAC assays, where even trace impurities can distort degradation kinetics or generate misleading biological signals. Our research- and screening-grade materials provide consistent performance across cell-based assays, biochemical studies, and SAR campaigns.
Complete Analytical Documentation (COA, MS, NMR)
Every batch is delivered with comprehensive analytical documentation, including Certificates of Analysis (COA), mass spectrometry (MS) data, and NMR spectra. This full data package ensures traceability, batch comparability, and regulatory readiness, supporting internal QC requirements, publications, and collaborative research projects. Researchers can confidently interpret assay results knowing that compound identity and purity are fully validated.
Advantage: Reproducibility, Accuracy, and Trusted Quality
Our high-purity pomalidomide derivatives are trusted by academic laboratories, CROs, and biotech companies worldwide for their reproducibility and analytical integrity. By eliminating variability introduced by poorly characterized reagents, we help researchers generate accurate, comparable, and publishable PROTAC data. This commitment to quality makes our products a dependable foundation for long-term protein degradation programs.
Strengthen PROTAC Assay Reliability with Verified High-Purity Reagents
If your research demands consistent degradation profiles and interpretable assay data, our high-purity pomalidomide derivatives provide the quality assurance your PROTAC workflows require. Contact us today to request technical documentation, samples, or a quotation and ensure your assays are built on trusted, analytically verified materials.
References
- Zattoni J, Vottero P, Carena G, et al. A comprehensive primer and review of PROTACs and their In Silico design[J]. Computer Methods and Programs in Biomedicine, 2025: 108687. https://doi.org/10.1016/j.cmpb.2025.108687.
- Vicente A T S, Moura S P S P, Salvador J A R. Synthesis, biological evaluation and clinical trials of Cereblon-based PROTACs[J]. Communications Chemistry, 2025, 8(1): 218. https://doi.org/10.1038/s42004-025-01598-9.
- Zhu Y, Dai Y, Tian Y. The Peptide PROTAC Modality: A New Strategy for Drug Discovery[J]. MedComm, 2025, 6(4): e70133. https://doi.org/10.1002/mco2.70133.
- Zattoni J, Vottero P, Carena G, et al. A comprehensive primer and review of PROTACs and their In Silico design[J]. Computer Methods and Programs in Biomedicine, 2025: 108687. https://doi.org/10.1016/j.cmpb.2025.108687.
- Distributed under Open Access license CC BY 4.0, without modification.
