Pomalidomide-based PROTACs are fast moving beyond hematological cancers to solid-tumor oncology and neurodegenerative disease. The cereblon-binding imide serves as a universal E3-ligase handle that can be tacked onto ligands for transcription factors, kinases or misfolded proteins; early pre-clinical studies demonstrate selective degradation of oncogenic drivers and toxic protein aggregates with prolonged pharmacodynamic effects that are unattainable with conventional inhibitors, positioning these degraders as next-generation therapeutics for protein-dysregulation driven diseases beyond those caused by enzymatic hyper-activity.
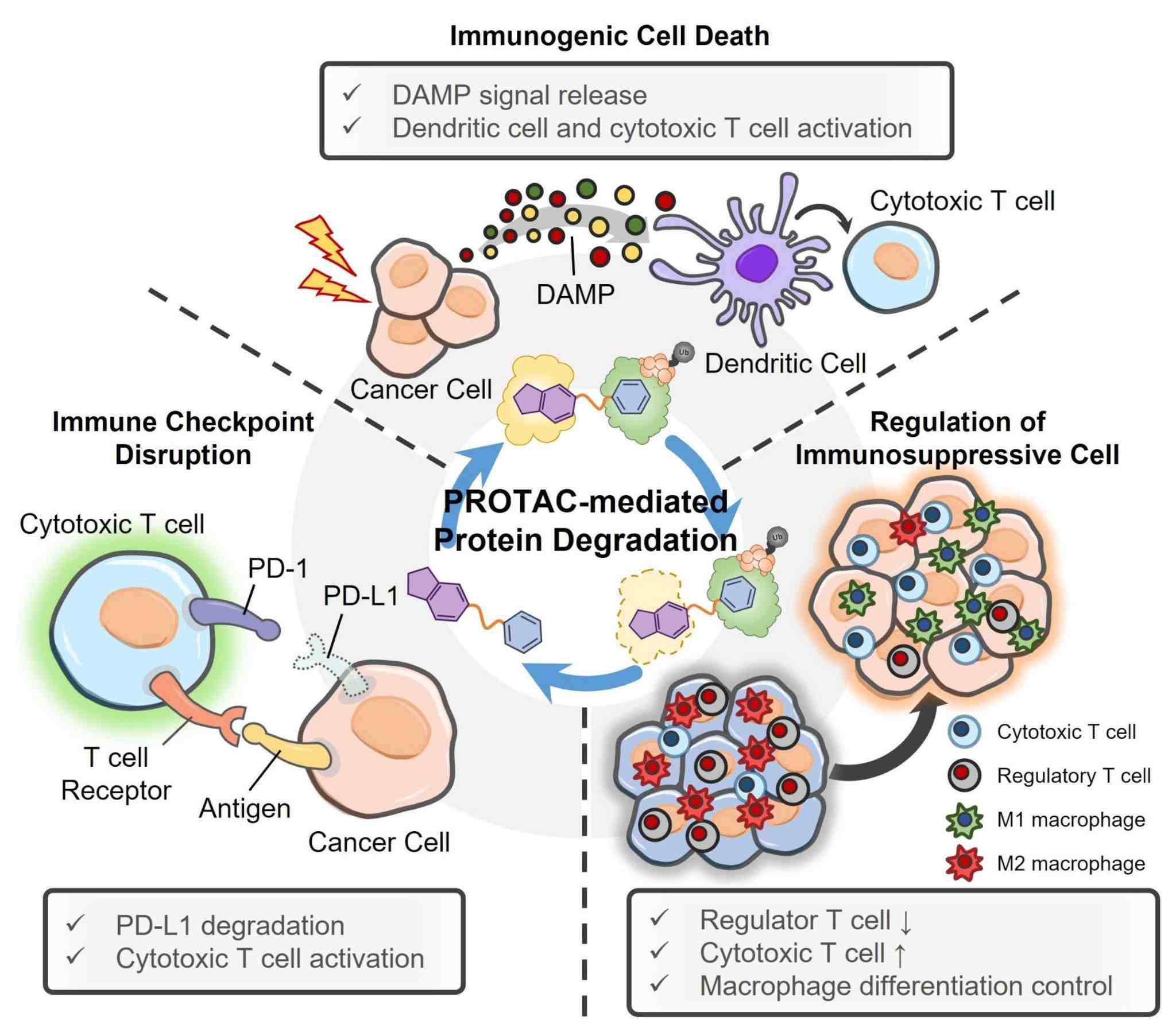 PROTAC-based cancer immunotherapy1,5
PROTAC-based cancer immunotherapy1,5
Introduction — The Expanding Scope of PROTAC Technology
PROTAC technology is no longer confined to the oncology space that gave it birth, and is today envisioned as a pan-therapeutic modality that is able to erase proteins that had been considered "undruggable" for lack of catalytic pockets or with shallow and transient interfaces. Because of the catalytic, event-driven nature of degradation, only transient binding is required, so scaffolding proteins, transcription factors and aggregation-prone neuroproteins are all legitimate targets. Pomalidomide-based prototypes are at the forefront of this expansion, as their CRBN-recruiting core is already qualified for human exposure, and optimization effort can focus on the linker and warhead, rather than the de-novo validation of the E3 ligase ligand.
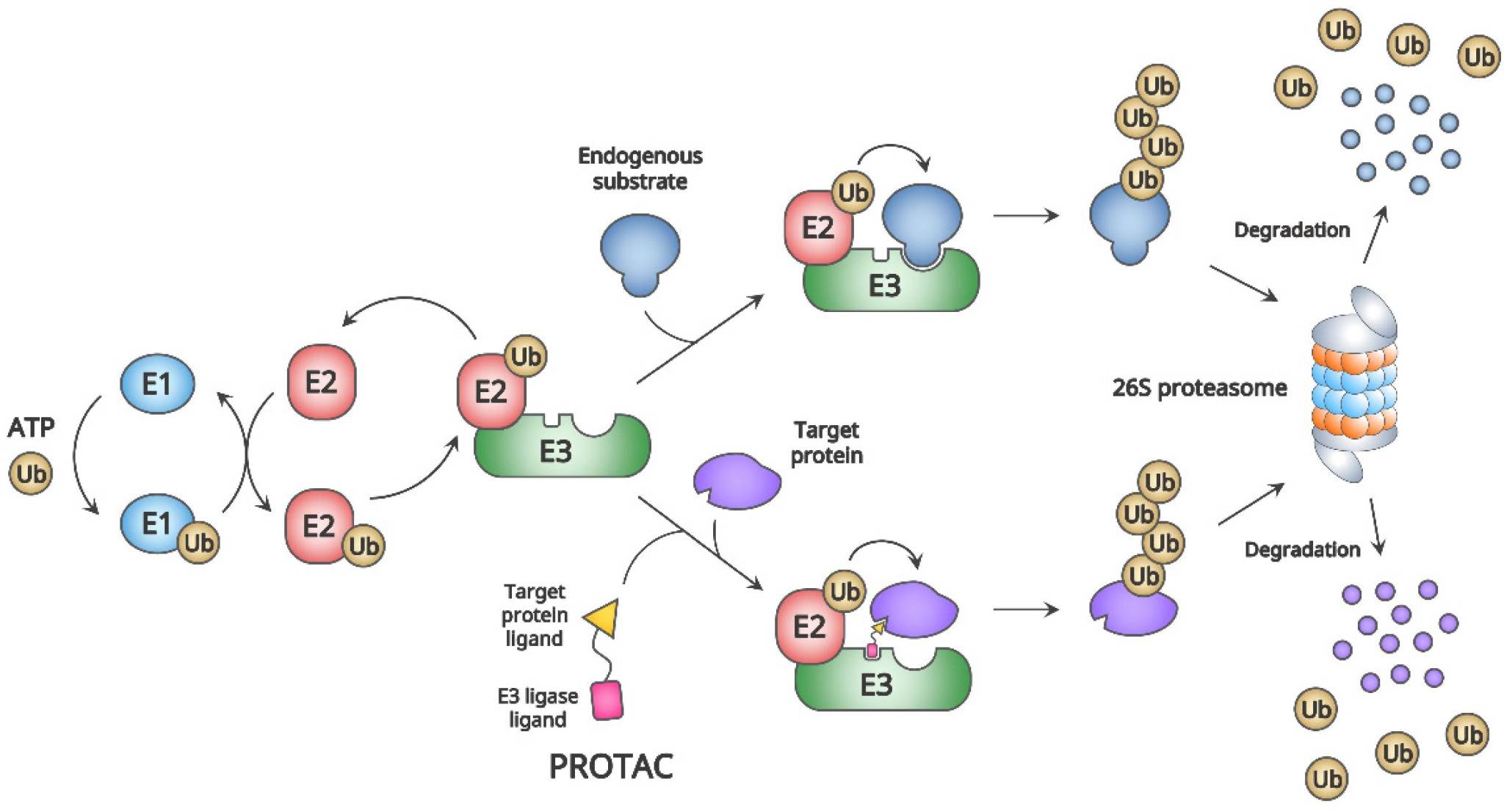 The ubiquitin–proteasome system (UPS) and proteolysis-targeting chimera (PROTAC)2,5
The ubiquitin–proteasome system (UPS) and proteolysis-targeting chimera (PROTAC)2,5
From Concept to Therapeutic Application
The conceptual shift – from blocking a protein to removing it – was initially described two decades ago with peptide-based recruiters, but it did not reach clinical development until small-molecule CRBN ligands like pomalidomide were fused into the chimera. The early clinical frontrunners have since validated the concept in hormone-sensitive cancers, and that same platform is now being redeployed to target drivers of neurodegeneration. The modular nature of these constructs means that the warhead can be swapped by medicinal chemists without changing the pomalidomide moiety, reducing discovery timelines and allowing medicinal chemists to draw upon prior safety readouts for the glutarimide backbone. In total, this translates to a shorter journey from proof-of-principle to first-in-human studies, and the regulatory baseline is more favorable than for entirely novel E3 recruiters, fast-tracking uptake across therapeutic areas.
Why PROTACs Represent a New Paradigm in Drug Discovery
Classical occupancy-based drugs need to achieve high systemic exposures to exert an inhibitory tone. This introduces the potential for dose-limiting toxicities and creates selection pressure for resistance through target over-expression. In contrast, PROTACs function catalytically, as a single degrader molecule can shuttle many substrates through the proteasome: efficacy is therefore achieved at lower exposures, and it can even persist after the compound has been cleared. The pomalidomide backbone further extends this advantage because its bicyclic core pre-organizes the ternary complex and minimizes the entropic cost of ubiquitin transfer, thus enabling degradation of low-abundance or high-affinity proteins that would otherwise compete with endogenous ligands. PROTACs therefore shift the drug-discovery metric from "how tightly can we bind?" to "how selectively can we recruit?" and opens up a target space that includes misfolded aggregates, transcriptional co-activators and scaffold proteins that are central to oncogenic and neurodegenerative signalling.
Role of Pomalidomide in Oncology-Focused PROTACs
Pomalidomide is a privileged CRBN-recruiting handle that transforms high-molecular-weight PROTACs into cell-permeant degraders without loss of catalytic turnover. Its bicyclic glutarimide contains a pre-organized bidentate carbonyl motif that intercalates into the zinc finger of CRBN, while the proximal phthaloyl ring provides a chemically silent perch for linker attachment. The dual bite of pomalidomide reduces the entropic cost of ternary-complex formation, enabling the degradation of low-abundance oncogenic proteins at exposures well below the immunomodulatory threshold of the parent IMiD.
Mechanism of CRBN Recruitment and Cancer Target Degradation
The PROTAC recruits pomalidomide into a hydrophobic pocket of CRBN, which allosterically changes the CRBN conformation to expose a neosubstrate interface into which the target protein is simultaneously captured by the distal ligand to form a ternary complex in which lysines on the oncogenic target are presented for ubiquitin transfer. The poly-ubiquitin chains are then elongated by the CUL4–DDB1–RBX1 complex, which channels the substrate into the 26 S proteasome for complete degradation. Pomalidomide is a true molecular glue, and thus does not require a pre-existing binding site on the target, which can be leveraged to eliminate "undruggable" transcription factors such as IKZF1/3, BRD4 or GSPT1 with nanomolar efficiency while leaving closely related paralogues that lack the critical lysine geometry unaffected.
Examples: BRD4, BCL-xL, and BTK PROTACs
dBET1 associates pomalidomide to a BET bromodomain ligand and mediates selective BRD4 degradation in leukaemia cell lines and delays tumor growth in xenograft models with oral dosing; compared to the parent BET inhibitor, the degrader mediates a more durable suppression of MYC transcription by removing both the catalytic and scaffolding functions of BRD4. Pomalidomide–BCL-xL PROTACs (e.g. DT2216) induce ubiquitination of the anti-apoptotic protein in solid-tumor cell lines and resensitise resistant clones to paclitaxel without affecting platelet viability (because platelets have low CRBN levels). BTK degraders such as NX-2127 combine pomalidomide with a covalent BTK binder and remove both wild-type and C481S mutant BTK in B-cell lymphoma models and restore sensitivity to PI3K inhibition, demonstrating how PROTACs can circumvent resistance mutations that inactivate conventional kinase inhibitors.
Table 1 Representative pomalidomide-based PROTACs in oncology research and development.
| PROTAC designation | Cancer target | Disease focus | Key advantage over inhibitor |
| dBET1 | BRD4 | AML, breast | Eliminates scaffolding function, longer MYC suppression |
| NX-2126 | BTK | B-cell lymphoma | Degrades C481S mutant, restores PI3K sensitivity |
| ARV-110 | AR | Prostate | Overcomes ligand-binding-domain mutations |
Synergy with Conventional Cancer Therapies
Since pomalidomide-based PROTACs deplete the protein instead of only inhibiting enzymatic activity, they have synergistic activity with chemotherapeutics that need an absence of prosurvival signalling to be effective. Following treatment with a BRD4 PROTAC followed by doxorubicin synergistically induces DNA-damage induced apoptosis in triple negative breast cancer spheroids, while co-treatment with a BCL-xL degrader and carboplatin resensitises ovarian cancer organoids to platinum induced cell death. The catalytic activity of PROTACs also allows for lower systemic exposure, therefore reducing the myelosuppression that is usually a limiting factor in combination regimens. The event-driven mechanism can also be effective even when the target protein is over-expressed or mutated, providing a therapeutic window that is impossible with occupancy based drugs.
Pomalidomide-Based PROTACs in Neurodegenerative Research
The metabolic characteristics of pomalidomide's glutarimide core allow for intermediate lipophilicity adjustment to enable delivery of bulky bifunctional payloads through the blood-brain barrier. As such, it is an attractive and active scaffold for targeting many previously undruggable proteins that have been implicated in ALS, Alzheimer's and Parkinson's disease. The causal proteins in these diseases are the product of toxic gain-of-function species that accumulate with time, so the catalytic activity of PROTACs enables chronic clearance even at sub-exposure levels and can thus avoid the dose-limiting toxicities that have limited traditional inhibitors to date. Recent proof-of-concept studies have demonstrated that pomalidomide-linked degraders can deplete pathogenic tau and α-synuclein aggregates in transgenic mouse models, without cross-reactivity against endogenous zinc-finger proteins, laying a framework for transition from onco-centric tool compounds to CNS-targeted therapeutics.
Degrading Pathogenic Proteins in ALS, Alzheimer's, and Parkinson's Models
In cell models of ALS, a pomalidomide-PROTAC recruiting CRBN to TDP-43 reduces cytoplasmic mis-localization and re-establishes nuclear import within hours, an outcome impossible to achieve with RNA-binding inhibitors that only suppress aggregation. For Alzheimer's disease, a degrader targeting hyper-phosphorylated tau eliminates both soluble and fibrillar species, resulting in reduced downstream phosphorylation of synaptic kinases and restoration of long-term potentiation in hippocampal slices. In Parkinson's cultures, an α-synuclein PROTAC reduces oligomeric seeds and restores dopamine uptake, while leaving intact the physiological pool of monomeric α-syn required for synaptic maintenance. In all three disorders the common mechanistic feature is the ability of the pomalidomide module to pre-organize a ternary complex inside neurons, enabling poly-ubiquitination of proteins that lack catalytic pockets and therefore are untargetable by traditional occupancy-based drugs.
Blood–Brain Barrier Penetration Challenges and Solutions
The challenge here is not poor affinity but efflux: the bicyclic phthalimide of pomalidomide is a moderate substrate for P-gp, which is expressed on the luminal membrane of brain capillaries, so that systemic exposure does not equate to parenchymal levels. Masking the C5 position with a small, non-polar spiro-ring reduces polar surface area without interfering with CRBN binding, moving the molecule out of P-gp recognition while maintaining passive diffusion. An alternative strategy is to embed a short, metabolically labile ester within the linker, creating a "soft" promoiety that is cleaved by brain esterases after entry, releasing the active degrader and trapping it behind the barrier. These design changes have produced pomalidomide-PROTACs whose unbound brain concentrations are higher than those of plasma-free drug, allowing for the degradation of intraneuronal targets at systemically administered doses that are well below the immunomodulatory ceiling for the parent IMiD.
Case Study: CRBN-Linked PROTACs in Tau and α-Synuclein Degradation
A degrader recently built by addition of a tau-recognizing heteroaryl to the C4 position of pomalidomide elicited near-complete clearance of pathogenic tau in rTg4510 mice following a single IP administration; phosphorylated tau species were reduced as early as six hours, with full suppression maintained over a seventy-two-hour period and corresponded to enhanced memory retention in contextual fear conditioning. In a similar α-synuclein model, a bumped pomalidomide analogue functionalized with a spiro-diazepine warhead elicited removal of detergent-insoluble α-syn from striatal homogenates with no discernible change in monomeric levels, a selectivity window not possible with synuclein antibodies, which cannot differentiate between the two. Off-target degradation of zinc-finger proteins was also minimized by steric "bumps" at the C5 position, confirming that judicious sculpting of the pomalidomide core can divorce the desired clearance of neuroproteins from IMiD-associated liabilities. These data position CRBN-linked degraders as a potential disease-modifying modality for proteinopathies that are otherwise bereft of curative options. A recent case study reports the design and characterization of a symmetrical pomalidomide–tau ligand homo-PROTAC that triggers CRBN-mediated self-ubiquitination and subsequent proteasomal degradation of CRBN itself, thereby limiting the available pool of ligase but still eliciting sufficient tau knock-down in primary cortical neurons. The inability of the negative control (N-methylated analogue, unable to bind CRBN) to decrease tau levels further indicates that the observed degradation was ligase-dependent, rather than a non-specific pharmacological effect. Western blot analysis revealed a selective loss of hyper-phosphorylated tau isoforms, with little change in total tau levels, indicating that only the pathologically modified species are recognized and ubiquitinated; a selectivity profile that is reminiscent of that seen with oncogenic neosubstrates, such as IKZF1/3.
Table 2 Neurodegenerative targets and delivery strategies for pomalidomide-based PROTACs.
| Disease | Target protein | Delivery route | Key outcome observed |
| Alzheimer's | Hyper-phosphorylated tau | Intracerebroventricular | ↓ p-Tau, ↑ synaptic markers |
| Parkinson's | α-Synuclein oligomers | Intranasal | Restored dopaminergic firing |
| ALS | Oxidized SOD1 mutants | Intrathecal | Extended survival, WT SOD1 spared |
| General | CRBN (self-degradation) | Systemic | Proof-of-concept for ligase control |
Translational and Preclinical Insights
Preclinical characterization of the pomalidomide based PROTACs is instructive and generally reflects a common theme. It is the glutarimide moiety which provides the 'pharmacologically qualified starting point', but the final therapeutic window is a function of distribution and linker-dependent off-target ZF degradation. Across a range of species, efficacy is achieved at a target exposure that is typically 1 log lower than the parent IMiD's IMID defining exposure, while observed toxicity is reversible lymphopenia and dose-limiting teratogenic events (as is known for CRBN engagement). These effects are preserved in a translation to human when adjusting plasma protein binding and hepatic clearance in a log/ALL model to allometrically scale for the human situation. Thus pomalidomide-PROTACs may provide a low-risk translational approach from preclinical proof of concept to FIH assessment.
In Vivo Efficacy and Toxicity Profiles
In orthotopic breast-cancer models, a pomalidomide-PROTAC targeting the estrogen receptor degrades the protein within 4 h and maintains suppression for 72 h, resulting in tumour stasis at oral doses that have no effect on peripheral white-cell counts. Off-target liabilities are dominated by IKZF1/3 depletion as evidenced by transient reductions in B- and T-cell subsets that recover within one week after cessation, recapitulating the lymphocyte dynamics of parental pomalidomide but at markedly lower Cmax. Hepatotoxicity is rare; elevation of transaminases is seen only when the linker exceeds a calculated aromatic density, above which hepatic clearance saturates. Reversibility is confirmed by a 28-day recovery phase in which all biochemical markers return to baseline despite continuous low-level exposure, indicating that the degrader is not causing cumulative tissue injury. In mouse xenograft models, oral dosing of CRBN-recruiting degraders results in ≥ 70 % knock-down of BRD4 or BTK within 6 h and suppression for > 24 h, correlating with maximal tumour growth inhibition without significant body-weight loss. Off-target degradation of GSPT1 is seen only at exposures ten-fold above the efficacious dose, indicating a therapeutic window that is wider than that of the parent IMiD. In neurodegeneration models, intracerebral delivery of tau-directed degraders lowers insoluble tau by half and restores synaptic marker expression without micro-haemorrhage or astrogliosis, supporting the feasibility of chronic CNS dosing. Plasma half-lives range from 2 to 5 h, yet pharmacodynamic effects persist for days, consistent with event-driven kinetics and slow protein re-synthesis rates.
Lessons from Current Academic and Biotech Studies
Experience from both academic consortia has shown that batch-to-batch variability in the diastereomeric composition is the best single predictor of in-vivo drift, with loss of ternary-complex potency by the circulating degrader and consequent target up-regulation when the C2 epimer of pomalidomide is above 0.5 %. Biotech-led programmes have solved this problem by using a crystallization seed that fixes the glutarimide in the desired configuration, leading to reproducible PK/PD between species. Early dog tox studies are another lesson shared by both sectors: the canine model can be used to predict human lymphocyte suppression with higher fidelity than rodents, and dose-finding can be refined before expensive primate studies. Both sectors also agree on using free-drug correction instead of total exposure for interspecies scaling, since the pomalidomide module is highly protein-bound but the pharmacodynamic read-out (target degradation) tracks the free fraction. Linker length and exit-vector geometry are critical: shortening aliphatic tethers from 12 to 6 atoms can double brain exposure without loss of ternary-complex stability, and replacing PEG with rigid bicyclic spacers to restrict motion decreases off-target neosubstrate recruitment. Academic groups have led the way on intranasal and focused-ultrasound delivery to circumvent the blood–brain barrier, while biotech efforts have focused on lyophilized powders reconstituted at the bedside to prevent hydrolytic degradation of the glutarimide ring. Early clinical read-outs show that resistance most often occurs through CRBN down-regulation rather than target mutation, leading to follow-up strategies that combine pomalidomide degraders with next-generation VHL or IAP E3 ligase ligands to retain degradation capacity in the relapse setting.
Our Pomalidomide Products and Research Support
Advancing PROTAC research in oncology and neurodegenerative diseases requires high-quality CRBN ligands, disease-focused design strategies, and reliable technical support. We provide a dedicated portfolio of pomalidomide-based products and research services to support protein degradation programs from early discovery through advanced preclinical evaluation, helping researchers translate mechanistic insights into actionable results.
Oncology-Grade Pomalidomide Derivatives and Screening Compounds
We supply a wide range of oncology-grade pomalidomide derivatives optimized for CRBN-mediated degradation of cancer-relevant targets such as BRD4, BTK, BCL-xL, and other oncogenic proteins. All compounds are manufactured to >98% purity and supported by full analytical documentation, ensuring consistency and reproducibility in cell-based and in vivo screening assays. Our ready-to-use screening compounds enable rapid evaluation of degradation potential across diverse cancer models.
Custom Design for CNS-Active PROTACs
For neurodegenerative disease research, we offer custom design services focused on developing CNS-compatible pomalidomide-based PROTACs. Our chemists optimize linker composition, lipophilicity, and molecular size to address blood–brain barrier penetration while maintaining effective CRBN recruitment. This tailored approach supports research targeting pathogenic proteins such as tau, α-synuclein, and other aggregation-prone substrates relevant to Alzheimer's, Parkinson's, and related disorders.
Advantage: Scientific Expertise, Proven Quality, and Global Support
Our partners benefit from deep expertise in IMiD chemistry, rigorous quality control standards, and responsive global supply capabilities. With direct access to experienced scientists, flexible order volumes, and reliable international logistics, we provide more than materials—we deliver end-to-end research support for complex PROTAC programs in oncology and neuroscience.
Advance Oncology and Neurodegeneration Research with Trusted PROTAC Solutions
Whether you are developing next-generation cancer degraders or exploring targeted protein removal in neurodegenerative disease models, our pomalidomide products and technical support are designed to move your research forward efficiently and reliably. Contact us today to discuss your project needs and access high-quality pomalidomide-based PROTAC solutions tailored to your therapeutic focus.
References
- Moon Y, Jeon S I, Shim M K, et al. Cancer-specific delivery of proteolysis-targeting chimeras (PROTACs) and their application to cancer immunotherapy[J]. Pharmaceutics, 2023, 15(2): 411. https://doi.org/10.3390/pharmaceutics15020411.
- Kim H, Park J, Kim J M. Targeted protein degradation to overcome resistance in cancer therapies: PROTAC and N-degron pathway[J]. Biomedicines, 2022, 10(9): 2100. https://doi.org/10.3390/biomedicines10092100.
- Vicente A T S, Moura S P S P, Salvador J A R. Synthesis, biological evaluation and clinical trials of Cereblon-based PROTACs[J]. Communications Chemistry, 2025, 8(1): 218. https://doi.org/10.1038/s42004-025-01598-9.
- Zhong G, Chang X, Xie W, et al. Targeted protein degradation: advances in drug discovery and clinical practice[J]. Signal transduction and targeted therapy, 2024, 9(1): 308. https://doi.org/10.1038/s41392-024-02004-x.
- Distributed under Open Access license CC BY 4.0, without modification.
