Pomalidomide-based PROTACs have emerged as one of the most versatile and widely adopted strategies in targeted protein degradation, offering powerful opportunities to redirect CRBN toward disease-relevant proteins. As degrader performance depends on much more than simple binding affinity, understanding the structure-activity relationships (SAR) behind pomalidomide modifications, linker engineering, and ternary complex formation has become essential for successful PROTAC design. This article provides a comprehensive, practice-oriented overview of how molecular architecture—ranging from C4/C5 substitution patterns to linker length, polarity, and stereochemistry—directly shapes degradation efficiency, selectivity, and drug-like behavior. By integrating mechanistic insights with real-world design strategies and case studies, we offer a clear roadmap for optimizing high-performance pomalidomide PROTACs and selecting the right building blocks to accelerate your discovery program.
Introduction—Why Structure-Activity Matters in PROTAC Chemistry
Proteolysis-targeting chimeras (PROTACs) have rapidly evolved from an academic concept to a transformative modality in drug discovery. Yet despite their conceptual elegance, successful PROTAC design is far from intuitive. Because these heterobifunctional molecules rely on orchestrating multiple molecular events—target engagement, E3 ligase recruitment, ternary complex formation, and ubiquitination—the structure-activity relationship (SAR) becomes the defining factor that determines whether a degrader is potent, selective, and developable.
In pomalidomide-based PROTACs, where cereblon (CRBN) serves as the recruited E3 ligase, subtle structural variations can dramatically influence biological outcomes. Even minor changes—such as shifting a substituent on the glutarimide ring or adjusting linker polarity—can alter ternary complex geometry, degradation kinetics, and off-target profiles. This sensitivity underscores a core principle in PROTAC chemistry: degradation efficiency is not solely a function of binding affinity, but of the precise spatial and energetic relationships between all three molecular partners.
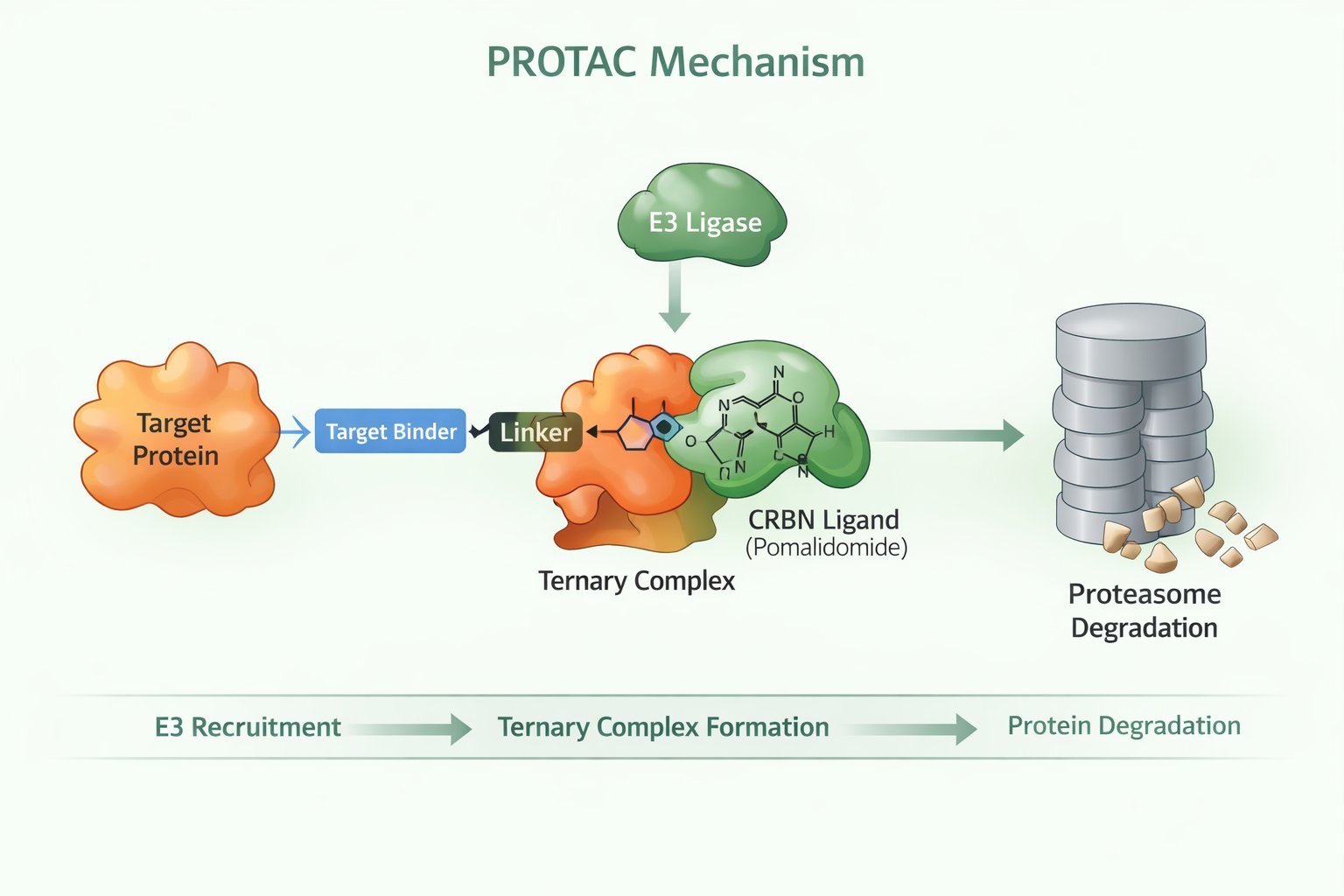 Fig 1. PROTAC mechanism
Fig 1. PROTAC mechanism
The Link Between Molecular Design and Degradation Efficiency
Unlike traditional inhibitors, PROTACs do not follow a simple "bind-and-block" mechanism. Their degradation activity depends on building a productive and stable ternary complex among the target protein, the PROTAC molecule, and CRBN. Therefore, molecular design choices directly dictate degradation performance, including:
- How efficiently the PROTAC recruits CRBN and the target simultaneously
- Whether the ternary complex geometry supports ubiquitin transfer
- How linker flexibility or steric hindrance affects productive protein-protein interactions
This is why empirical optimization—guided by rational SAR analysis—is indispensable. A PROTAC with excellent binary binding can still fail to degrade if its spatial conformation is not conducive to ternary assembly.
How CRBN-Binding Geometry Shapes Biological Outcomes
Pomalidomide, a clinically validated immunomodulatory drug (IMiD), is one of the most widely used CRBN ligands in PROTAC design due to its favorable binding orientation and modifiable exit vectors. However, modifications to the phthalimide core or glutarimide ring can shift CRBN-binding geometry, influencing:
- Degradation selectivity, especially toward neo-substrates like IKZF1/3
- Off-target liabilities, which often stem from unintended CRBN engagement behaviors
- Overall degrader potency, as altered geometry can disrupt cooperative interactions within the ternary complex
Understanding these structure-activity principles allows chemists to fine-tune pomalidomide-based PROTACs for optimized degradation profiles while minimizing unwanted biological effects.
Core Structural Components of Pomalidomide-Based PROTACs
Pomalidomide-based PROTACs rely on a modular architecture that allows chemists to precisely tune degradation performance. Each structural element—the CRBN-recruiting ligand, the target-binding moiety, and the linker—contributes distinct functional and biophysical properties. Understanding how these components interact is essential for rational PROTAC design and for achieving efficient, selective protein degradation.
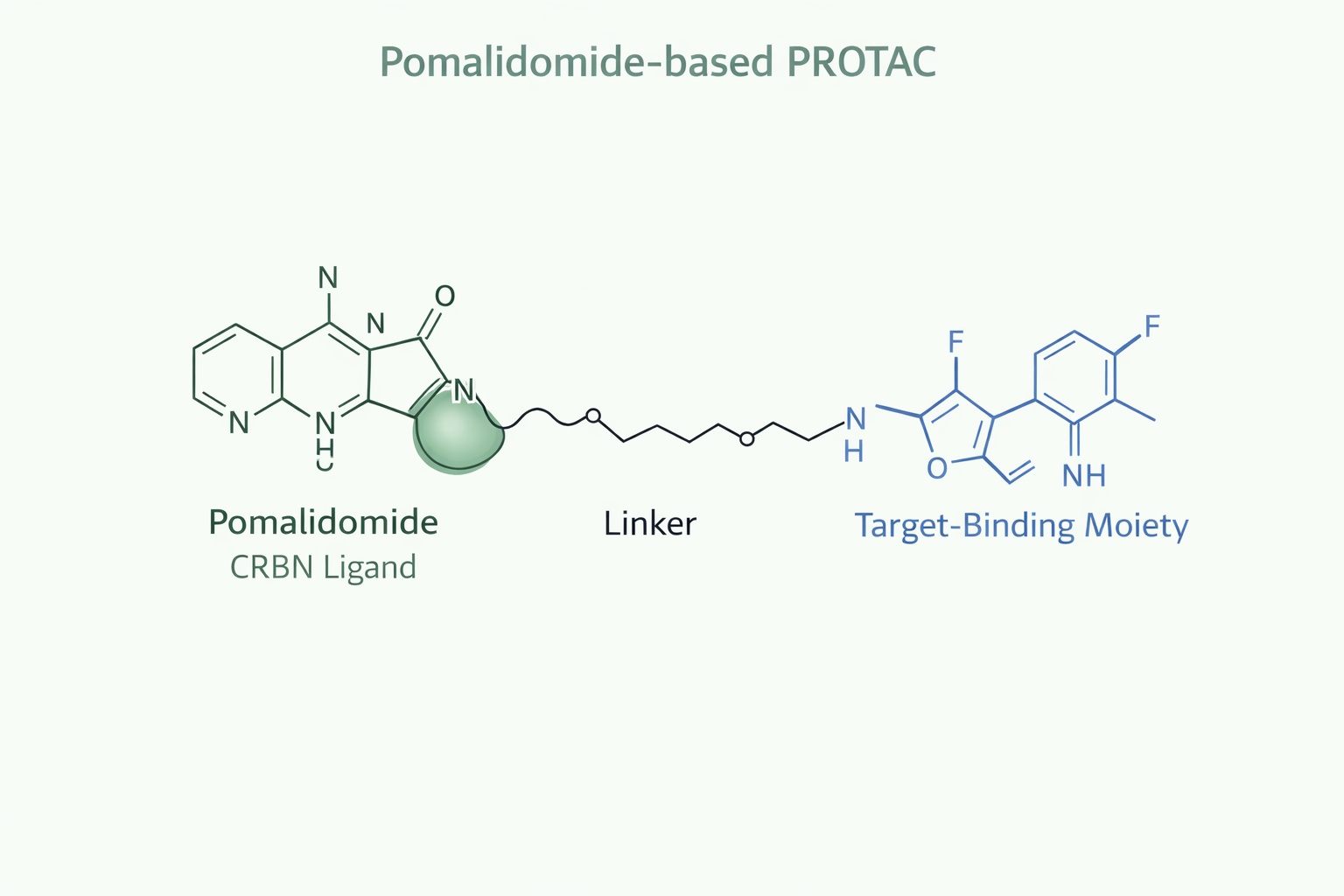 Fig 2. A detailed schematic diagram of a pomalidomide-based PROTAC
Fig 2. A detailed schematic diagram of a pomalidomide-based PROTAC
Pomalidomide serves as a versatile CRBN ligand because of its:
- High-affinity binding to the CRBN thalidomide-binding domain
- Well-characterized exit vectors at the C4 and C5 positions
- Predictable behavior in ternary complex formation
Chemical modifications around the phthalimide ring and glutarimide scaffold enable fine control over CRBN recruitment and substrate selectivity. For example:
- C4 functionalization typically preserves strong CRBN binding and provides a robust attachment point for linkers.
- C5 substitutions can shift CRBN-binding orientation, modulating ternary complex cooperativity and reducing off-target degradation of neo-substrates like IKZF1/3.
These structure-activity insights are foundational for designing PROTACs with optimal degradation windows and minimal adverse effects.
Target Binder and Linker Integration
The target-binding ligand dictates biological specificity, but its integration with the pomalidomide warhead determines whether the PROTAC can simultaneously engage both proteins. Key considerations include:
- Binding orientation: Proper spatial presentation is required to support ternary complex stabilization.
- Exit vector compatibility: Misaligned attachment points can disrupt target engagement even if both ligands bind well individually.
- Physicochemical balance: Lipophilicity, hydrogen-bonding potential, and steric footprint all influence cellular permeability and degradation kinetics.
A successful PROTAC must balance these factors so that its target ligand and CRBN ligand operate cooperatively—not competitively—within the assembled complex.
Ternary Complex Assembly and Stability
The hallmark of PROTAC pharmacology is the formation of a stable, cooperative ternary complex. Its quality depends on:
- Protein-PROTAC-protein interface complementarity
- Favorable enthalpic and entropic contributions
- Reduced steric clashes between the two proteins
Even PROTACs with strong binary affinities can fail if their ternary geometry is suboptimal. Conversely, weak binders can become potent degraders when cooperative stabilization occurs. Thus, ternary complex formation is not merely a downstream event—it is a central design target, deeply influenced by every structural decision in the PROTAC molecule.
Key Structure-Activity Relationships (SAR)
Understanding the structure-activity relationships (SAR) of pomalidomide-based PROTACs is essential for designing degraders that exhibit strong potency, favorable selectivity, and predictable pharmacological outcomes. Because PROTACs function through cooperative ternary complex formation rather than simple binary binding, their SAR landscape is intricate. Substituent positioning, linker architecture, stereochemical configuration, and overall molecular conformation all contribute to a complex interplay that ultimately governs degradation efficiency.
Modification at C4 and C5 Positions — Impact on Binding Affinity
The pomalidomide scaffold offers two primary vectors for structural modification: C4 and C5, each with distinct SAR implications.
C4 Modifications:
- Widely considered the most reliable attachment point for linker installation.
- Typically maintain strong CRBN binding affinity and predictable ligand orientation.
- Provide a stable geometry for consistent ternary complex assembly across diverse target classes.
C5 Modifications:
- Impact CRBN binding orientation more dramatically.
- Can fine-tune neo-substrate recruitment, potentially reducing off-target degradation of IKZF1/3.
- Require more empirical testing due to potential disruptions in ternary complex cooperativity.
From a SAR perspective, C4 functionalization is generally preferred for early PROTAC screening, while C5 derivatives are ideal for advanced optimization when selectivity or off-target risk becomes a priority.
Effect of Linker Length and Polarity on Target Engagement
The linker is often described as the "tuning dial" of PROTAC molecules. Its length, rigidity, polarity, and attachment geometry directly affect how the target protein and CRBN are positioned relative to each other. Key SAR insights include:
- Short linkers may limit flexibility, preventing optimal protein-protein alignment and reducing degradation efficiency.
- Overly long or flexible linkers can introduce entropic penalties, decreasing ternary complex stability.
- Polar linkers (e.g., PEG-based) improve solubility but may reduce membrane permeability.
- Hydrophobic linkers enhance cell penetration but may increase nonspecific binding or metabolic liabilities.
In practice, medicinal chemists often explore linker "length series" to identify the sweet spot where ternary complex formation is maximized without compromising physicochemical properties.
| Linker Parameter | Effect When Increased | Effect When Decreased | Practical Design Guidance |
| Linker Length | Improves spatial reach and flexibility but may reduce ternary complex cooperativity due to entropic penalties | Enhances rigidity and cooperativity but may limit productive protein-protein alignment | Evaluate a length series to identify the optimal balance for ternary complex formation |
| Linker Polarity | Increases aqueous solubility but may compromise cell permeability | Improves membrane permeability but may increase nonspecific binding or aggregation | Combine polar and hydrophobic segments to balance solubility and permeability |
| Flexibility | Allows adaptive binding but can decrease ternary complex stability | Promotes defined geometry and cooperative binding | Use flexible linkers for dynamic targets; introduce rigidity during optimization |
| Rigidity | Pre-organizes PROTAC conformation and reduces entropic cost | May cause steric clashes or misalignment | Incorporate aromatic, alkyne, or cyclic motifs selectively |
| Exit Vector Geometry | Enables favorable protein orientation if aligned correctly | Misalignment can disrupt ternary complex formation | Optimize attachment points on both ligands early in design |
| Steric Bulk | Can enhance selectivity by discouraging off-target complexes | Excess bulk may reduce binding efficiency | Introduce steric elements strategically to tune selectivity |
Influence of Stereochemistry and Conformation
Stereochemistry—both in the linker and the target ligand—plays a critical role in PROTAC performance. Because ternary complex assembly is highly three-dimensional, even a single stereocenter can determine whether the target and CRBN adopt a compatible orientation. Important SAR observations:
- Stereochemical mismatches can prevent ternary complexization despite strong binary affinities.
- Rigid linkers or conformational constraints can pre-organize the PROTAC into productive geometries, reducing entropic barriers to complex formation.
- Flexible linkers, while easier to synthesize, may create heterogeneous conformations that hinder cooperative binding.
Advanced degrader design often introduces conformational control elements—such as spiro, cyclopropyl, or aromatic linkers—to lock the PROTAC into more favorable orientations.
Practical Optimization Strategies
Designing a high-performance pomalidomide-based PROTAC requires more than ligand selection and linker attachment—it demands iterative optimization based on mechanistic insight, SAR learnings, and empirical degradation data. The strategies below reflect industry best practices used by leading PROTAC developers to refine degrader potency, selectivity, and drug-like properties.
Reducing Off-Target Degradation While Preserving Activity
One of the recurring challenges in CRBN-based degrader design is minimizing unintended modulation of CRBN neo-substrates such as IKZF1/3. These off-target effects can complicate biological readouts and introduce clinical risks. Effective mitigation strategies include:
- C5-modified pomalidomide derivatives: Adjustments at the C5 position can alter CRBN's recruitment surface, reducing the propensity to degrade endogenous substrates while preserving target-directed degradation.
- Linker-induced geometric tuning: Introducing steric bulk or modifying linker exit geometry can shift ternary complex formation away from CRBN neo-substrate conformations.
- Fine-tuning CRBN binding affinity: Slight reductions in affinity can sometimes reduce off-target activity without compromising overall PROTAC potency, especially when ternary cooperativity is strong.
- Target-ligand-driven selectivity: High target engagement affinity can improve target bias by favoring the intended ternary complex configuration.
Balancing these parameters ensures that pomalidomide-based PROTACs deliver efficient degradation while maintaining a cleaner biological profile.
Adjusting Linker Composition for Optimal Degrader Efficiency
- Linker engineering remains the most powerful and flexible axis of PROTAC optimization. Several practical adjustments consistently improve performance:
- Length Scanning: A systematic approach where multiple linker lengths are synthesized to identify the most cooperative ternary geometry.
- Rigid vs. Flexible Linkers:
Rigid linkers (aryl, alkyne, piperazine, spiro motifs) can pre-organize the molecule into favorable orientations.
Flexible linkers (PEG, alkyl chains) provide adaptability for targets with dynamic surfaces.
- Polarity Tuning: Blending hydrophobic and hydrophilic segments maintains solubility while enhancing cell permeability.
- Exit Vector Optimization: Ensuring the linker leaves the pomalidomide core at a precise trajectory is essential for assembling a productive ternary complex.
In practice, this involves generating a linker matrix—varying length, polarity, rigidity, and attachment vectors—to map the degradation landscape for each target protein.
Case Studies: HDAC, BTK, and BRAF PROTACs
Real-world examples illustrate how SAR and optimization principles translate into successful degrader design:
HDAC PROTACs
HDACs present multiple isoforms and complex binding pockets, making linker geometry crucial. Pomalidomide-based HDAC degraders typically benefit from:
- Medium-length PEG linkers for flexible positioning
- C4 pomalidomide attachment for consistent CRBN orientation
- Rigid aromatic elements to enhance ternary cooperativity
These design choices often result in selective HDAC6 degradation with minimized systemic effects.
BTK PROTACs
BTK's compact ATP-binding pocket demands precise target-ligand orientation. Successful BTK degraders often incorporate:
- C4-linked pomalidomide for stable CRBN binding
- Short to medium aliphatic linkers that avoid steric clashes
- Conformationally biased linkers that align BTK with CRBN binding surfaces
These features collectively improve potency and reduce reliance on covalent binding strategies.
BRAF PROTACs
BRAF's large, flexible kinase domain benefits from PROTAC-induced proximity effects. Effective BRAF degraders commonly employ:
- Longer linkers to span the distance between CRBN and BRAF surfaces
- Mixed-polarity linkers to maintain permeability
- Stereochemically controlled target ligands to ensure orientation fidelity
This enables efficient degradation of wild-type and mutant BRAF variants.
| Target Protein | Preferred Linker Characteristics | Pomalidomide Attachment Site | Key SAR Considerations | Typical Application Scenarios |
| HDAC | Medium-length, flexible PEG or mixed-polarity linkers | C4 | Requires sufficient conformational freedom to accommodate isoform diversity and surface dynamics | Selective degradation of HDAC6 and related epigenetic targets |
| BTK | Short to medium-length linkers with moderate rigidity | C4 | Precise geometric alignment needed to avoid steric interference with the ATP-binding pocket | Degradation of wild-type and mutant BTK, including resistance variants |
| BRAF | Longer linkers with balanced polarity | C4 or C5 | Large kinase domain benefits from extended reach and careful control of linker orientation | Targeted degradation of BRAF WT and oncogenic mutants (e.g., V600E) |
Our Pomalidomide-Based PROTAC Solutions
As PROTAC design becomes increasingly sophisticated, researchers need reliable, high-quality building blocks and expert support to accelerate discovery timelines. Our pomalidomide-based PROTAC solutions combine validated chemistry, scalable synthesis, and deep SAR expertise to help you design degraders with superior potency, selectivity, and developability.
SAR-Validated CRBN Ligands Ready for Customization
We offer a curated portfolio of pomalidomide-derived CRBN ligands engineered for high-performance PROTAC construction. Each building block is designed with medicinal chemistry and SAR principles in mind:
- C4- and C5-functionalized pomalidomide derivatives for flexible exit vectors
- Linker-ready handles (alkyne, azide, amine, carboxyl, PEG variants) enabling seamless conjugation
- Selectivity-tuned analogs to reduce off-target degradation (e.g., IKZF1/3 sparing)
- High-purity, research-grade materials optimized for library synthesis and lead optimization
Whether you are exploring early ternary complex feasibility or refining late-stage degrader candidates, our ligands provide a robust foundation for successful CRBN-based PROTAC design.
Expert Support in Linker Design and Derivative Synthesis
PROTAC optimization is rarely linear—and that's where our scientific team adds value. We provide strategic guidance and tailored solutions across the molecular design workflow:
- SAR-driven linker recommendations based on your target class and structural constraints
- Custom synthesis of novel pomalidomide derivatives for advanced mechanistic or selectivity studies
- Rapid production of linker series to map optimal degrader geometry
- Consultative support on ternary complex modeling, physicochemical balancing, and off-target risk mitigation
Our chemists and PROTAC specialists work collaboratively with your team to accelerate decision-making and reduce costly synthesis cycles.
Advantage: Proven Chemistry, Fast Delivery, and Technical Consulting
Your PROTAC pipeline deserves more than off-the-shelf reagents. We deliver strategic advantages that elevate your discovery program:
- Proven synthetic routes that ensure batch-to-batch consistency
- Expedited delivery timelines for rapid iteration and parallel testing
- Application-focused technical consulting to help you interpret SAR trends and refine degrader design
- Flexible engagement models, from single-compound orders to full custom synthesis packages
With our integrated approach, you gain not just high-quality pomalidomide building blocks, but a reliable scientific partner committed to your success in targeted protein degradation.
Get Expert Support for Pomalidomide PROTAC Design and Custom Synthesis
Advancing a pomalidomide-based PROTAC from concept to optimized degrader requires precise structural engineering, validated CRBN ligands, and informed SAR decision-making. Whether you're building your first degrader series or refining a lead compound, our team provides the scientific expertise and custom chemistry support needed to accelerate your program. Contact us to receive a tailored consultation and actionable recommendations for your specific target and discovery objectives.
