PROTAC targeting HMGCR
HMGCR is a lipid metabolism enzyme located in the endoplasmic reticulum (ER) with a transmembrane domain at the N terminus and a catalytic domain at the C terminus that converts HMG-CoA to mevalonate, the precursor of cholesterol. As the rate-limiting enzyme for cholesterol synthesis, HMGCR expression under physiological conditions is highly regulated by transcription, post-transcription, translation, and post-translation.
A large number of studies have shown that the increase of HMGCR level will promote the accumulation of cholesterol, leading to the increased risk of hypercholesterolemia, while the inhibition of HMGCR will reduce plasma LDL cholesterol and effectively prevent atherosclerosis. Therefore, HMGCR is an important target for the treatment of disorders of lipid metabolism. Statins are the most widely used class of cholesterol-lowering drugs, which play a competitive role by occupying part of the HMG-CoA binding site and eventually reducing the serum cholesterol level of patients with hypercholesterolemia.
However, considering the drug resistance and serious side effects, researchers are still looking for safer and more effective treatment methods targeting HMGCR, including PROTAC technology.
Luo's group first designed and synthesized a series of PROTACs targeting HMGCR. They conjugated atorvastatin to the CRBN ligand pomalidomide by the linker and employed different lengths of the linker to determine whether they affected the effect of PROTAC. SAR studies of this compound series showed that the linker length seems to be very important for the degradation efficacy of PROTAC, and PROTAC can induce significant degradation of HMGCR when the linker length reaches 22 atoms or more. Among them, P22A with a DC50 value of 0.1 μM showed the strongest pro-degradation effect on endogenous HMGCR.
It is worth mentioning that P22A was shown to be effective in reducing HMGCR protein levels and blocking cholesterol biosynthesis in Huh7 cells without triggering a compensatory upregulation of HMGCR. This study makes HMGCR the first ER-localized multimer transmembrane protein that can be degraded by PROTAC technology, confirming the feasibility of PROTAC technology for the treatment of the metabolic syndrome.
In the work published in 2021, Zhu's research group designed PROTAC with lovastatin acid as the target and further screened the types of the linker and E3 ubiquitin ligands. PROTAC using VHL or CRBN as E3 ligase ligands exhibited potent degradation of HMGCR. Unlike most PROTACs, which have poor in vivo activity and PK metrics, the VHL ligand-based HMGCR-PROTACs (21b and 21c) identified by the team have good oral bioavailability and demonstrated efficacy in a mouse model of hypercholesterolemia. A first example of PROTAC targeting by oral HMGCR was provided.
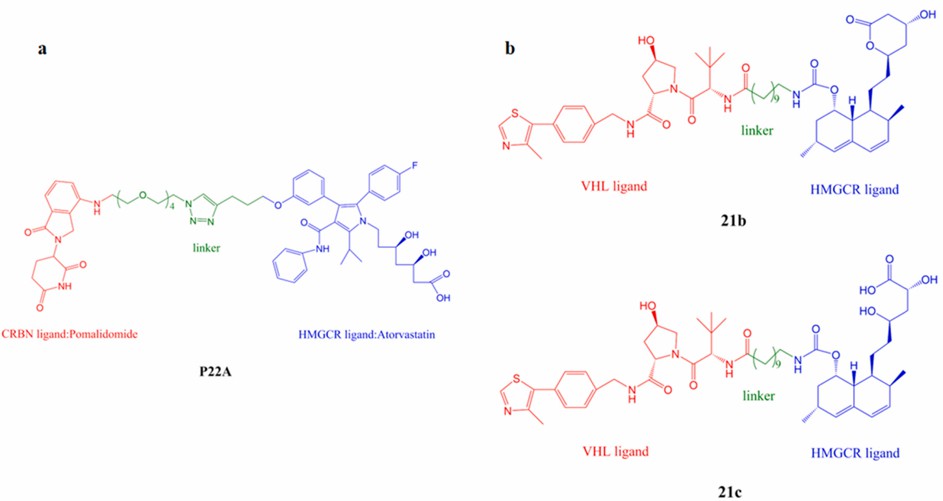 Fig 1. PROCTAC molecules targeting HMGCR
Fig 1. PROCTAC molecules targeting HMGCR
PROTAC targeting PNPLA3
"A 481-amino acid protein encoded by the Patatin phospholipase domain containing 3(PNPLA3) gene is an enzyme with lipase activity toward both triglycerides and retinyl esters and acyltransferase activity toward phospholipids." Fatty liver disease (FLD) is a condition associated with genetic, environmental, and metabolic stress that leads to inflammation, fibrosis, and cirrhosis. Proteomics and metabolomics studies suggest that its pathogenesis may be related to reduced lipid turnover and LD release. PNPLA3 variant 148 M is a key factor in the induction of fatty liver, which helps the enzyme escape ubiquitin-mediated degradation and accumulate on lipid droplets, eventually leading to fatty liver.
At present, the mechanism of PNPLA3-induced liver disease has not been fully elucidated, but its activity and expression level do affect liver lipid metabolism. Degradation and depletion of PNPLA3 is a potential therapeutic intervention strategy. Basu's group designed and synthesized a PNPLA3 degradation product (PROTAC3), which contains a hydroxyproline derivative with a high affinity for E3 ligase VHL.
In the biological experiments, PROTAC3 showed a concentration-dependent degradation effect on QBI-293A cells expressing PNPLA3. In addition, they also tested the role of PROTAC3 in mice expressing PNPLA3(WT) and PNPLA3(148 M), verifying that PROTAC technology can reduce PNPLA3(148 M) levels and improve FLD related to mutant protein expression. Unfortunately, the structure of PROTAC3 is not publicly available. This study confirms that some fatty liver diseases are related to the accumulation of PNPLA3 in lipid droplets, and also provides a new idea for the treatment of fatty liver.
PROTAC targeting LXR
Liver X receptors α and β (LXRα and LXRβ) belong to the nuclear hormone receptor superfamily and are ligand-dependent transcription factors that play regulatory roles in cholesterol homeostasis and lipid homeostasis. LXR is ubiquitously expressed in a variety of tissues and is associated with a variety of pathological conditions such as atherosclerosis, diabetes, and obesity. Therefore, targeting LXR proteins has been the focus of research in the development of cholesterol-lowering drugs and the treatment of atherosclerosis, such as the development of LXR agonists, antagonists, and reverse agonists.
However, the poor development of LXR inhibitors due to their weak binding activity and difficulty in synthesis and preparation has limited the clinical application of LXR inhibitors and led scientists to search for new inhibitory tools to act on this protein.
By referring to the X-ray crystal structure information of LXRα and its agonist GW3965, Tsuji's group determined the linker position of LXRα, thus successfully developed a series of E3 ligands containing different E3 ligands (CRBN binding pomalidomide; VH032 binding VHL) and PROTACs with different linker lengths (PEG3, PEG4, PEG5, and PEG6).
The results of SAR studies showed that PROTAC containing PEG5 and VHL recruitment (GW3965-PEG5-VH032) showed potent degradation activity against LXRβ, and a hook effect of PROTAC at high concentrations was also found (Fig 2). This study provides new therapeutic strategies for LXR-based related metabolic diseases.
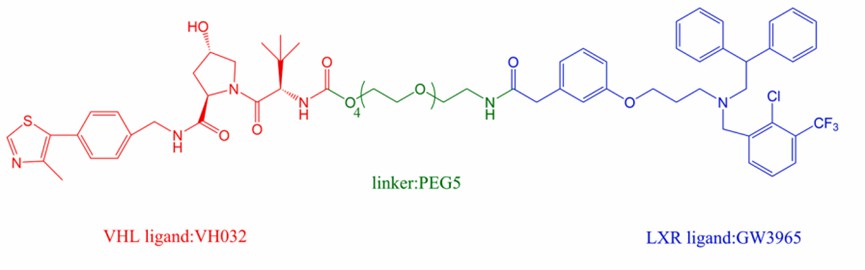 Fig 2. The structure of PROTAC targeting LXR
Fig 2. The structure of PROTAC targeting LXR
Phosphatidylinositol 3-kinase (PI3K) is a lipid kinase involved in intracellular signaling and plays an integral role in regulating many different cellular functions, including cell growth, proliferation, differentiation, and transcription and translation. Dysregulation of PI3K activity as well as aberrant PI3K signaling has been detected in many diseases, such as type 2 diabetes and cardiovascular disease. Therefore, targeting PI3K is becoming a promising therapeutic strategy for metabolic diseases.
However, intrinsic and acquired drug resistance and possible toxicity of PI3K inhibitors have limited their clinical application. With the rapid development of targeted protein degradation technology, some researchers have tried to apply PROTACs to the degradation of PI3K. Based on the CRBN E3 ligase ligands pomalidomide and ZSTK474, Li's group designed a series of new (B, D, E, F) targeted for the degradation of PI3K (Fig 3). Four compounds were shown to induce significant degradation of PI3K and reduce phosphorylation of Akt, 70S6K, and GSK-3β in hepatoma HepG2 cells. All four PROTAC compounds exhibited good enzymatic activity and showed degradation of PI3K proteins in a time - and concentration-dependent manner.
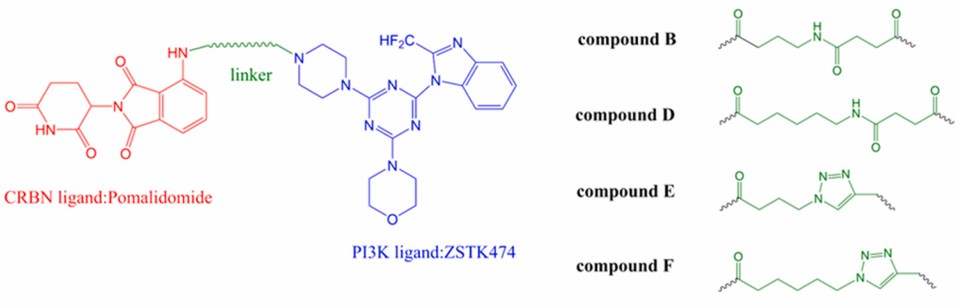 Fig 3. The structure of PROTAC targeting PI3K
Fig 3. The structure of PROTAC targeting PI3K
By recruiting VHL E3, Liu's group obtained a series of PROTACs degraders HL-4/5 and HL-6 to HL-9 containing VHL ligands to target PI3K. Subsequent biological activity studies confirmed that most PROTACs, especially HL-8, can significantly degrade PI3K kinase at a certain dose. Although the degradability of PROTAC molecules has not been confirmed in metabolic syndrome phenotypes, in vitro experiments confirmed the feasibility of applying PROTAC technology to PI3K kinase, providing a new way for drug design targeting PI3K.
PROTAC targeting PDEδ
Isoprene molecular chaperone (PDEδ), a lipid-binding protein that forms a diffusible complex with isopentenylated OS proteins and provides a plasma membrane localization function for isopentenylated Ras proteins, is generally considered a potential cancer therapeutic target. Based on the benzyl derivative of the PDEδ inhibitor deltasonamide 1 as the target head, Winzker's group selected pomalidomide (a CRBN ligand) and cullin 2 (a VHL ligand) as E3 ligase ligands, respectively. And by linking them into chimeras with three different ethylene glycol unit length linkers, a series of PROTACs capable of efficiently degrading PDEδ were obtained (Fig 4).
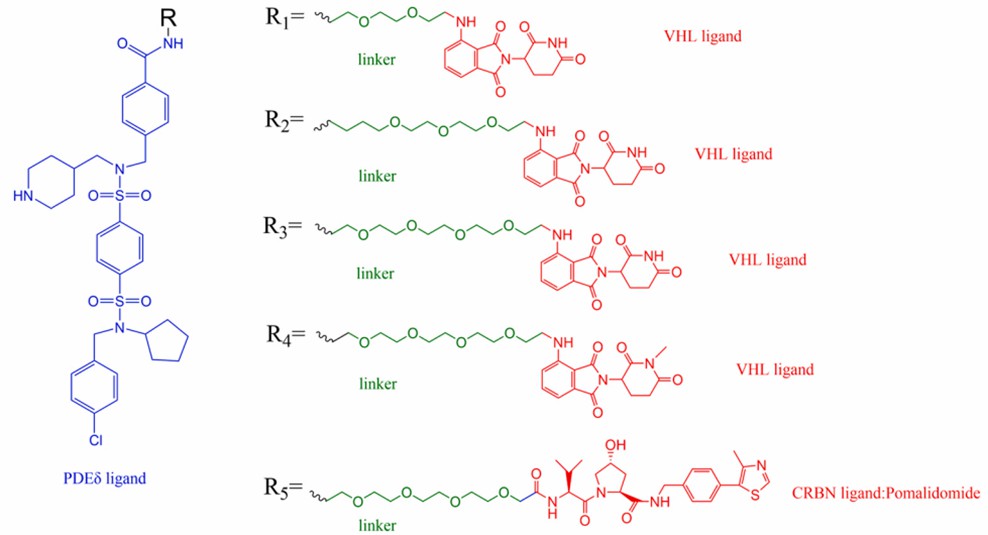 Fig 4. The structure of PROTAC targeting PDEδ
Fig 4. The structure of PROTAC targeting PDEδ
The biological results fully demonstrated that these PROTACs can effectively and selectively down-regulate the level of PDEδ in pancreatic cancer cell lines. "However, several proteins were expressed in PROTAC-treated cells in higher abundance than in DMSO-treated controls, particularly enzymes involved in the mevalonate pathway, which is associated with lipid metabolism." These findings suggest that PROTAC-mediated degradation of PDEδ may enhance the expression and activity of sterol regulatory element-binding protein (SREBP), which in turn stimulates the expression of several enzymes involved in lipid metabolism, leading to the elevation of cholesterol precursor levels.
Although PROTAC in this study did not target specific metabolic-related proteins for degradation, this finding provides the first evidence that PDEδ plays a role in the regulation of cholesterol synthesis and provides more insights into the regulation of pathogenic proteins related to metabolic syndrome.
Estrogen-related receptor α (ERRα) is a member of ERR family (including ERRα, ERRβ, and ERRγ), which is an orphan nuclear receptor (NR). Studies have shown the role of ERRα in the maintenance of mitochondrial homeostasis, energy metabolism, adaptive thermogenesis, and adipogenesis. The change in ERRα levels may be related to the long-term development of various metabolic disorders, such as osteoporosis, obesity, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), insulin resistance, type 2 diabetes mellitus, and cardiovascular disease. Therefore, ERRα is considered to be a popular target for the treatment of metabolic syndrome.
Carnosic acid (CA) is a phenolic diterpenoid compound with antioxidant and anti-aging effects and is commonly used to treat obesity and cardiovascular diseases. Zheng's group found that CA could inhibit RANKL-induced osteoclast formation and improve OVX-induced bone loss by simultaneously targeting SREBP2 and ERRα. CA was also found to possess the properties of molecular glue, which further enhances the interaction between ERRα and E3 ubiquitin after binding to the ligand-binding domain of ERRα to promote its ubiquitination for proteasomal degradation.
In conclusion, CA has a specific inhibitory effect on cholesterol metabolism in a dual-target targeted inhibition manner, providing a representative example of molecular glue intervention in physiological metabolism.
Molecular glue targeting SCAP
SREBP cleavage activating protein (SCAP) is a key regulatory protein in cholesterol metabolism. It binds to SREBP to form the SCAP-SREBP complex, which is transported from the ER to the Golgi for proteolysis and release of the SREBP transcription factor domain, which then enters the nucleus to induce synthesis and uptake of fatty acids, TG, and cholesterol.
Studies have shown that specific SCAP knockdown may reduce lipid accumulation and intracellular oxidative stress, and promote lipid clearance by enhancing lipid autophagy, thereby reducing atherosclerotic plaque formation. On the contrary, overexpression of SCAP may enhance SREBP nuclear translocation and lead to the accumulation of various lipids and ultimately pathogenicity. Therefore, targeted SCAP inhibition may be an important strategy for the treatment of metabolic syndrome. Lycorine is an alkaloid, isolated from Lycoris, which has various pharmacological activities such as anti-inflammatory, antifungal, antiviral, anti-malarial, and anti-tumor.
Zheng's group found lycorine, a molecule that can target SCAP, through high-throughput screening in vitro. The effect of lycorine on reducing fat, serum TC and TG levels in vivo was verified in a HFD mouse model. Mechanistically, lycorine, as a MG, interacts with both SCAP and sequentosome 1 (SQSTM1) /p62 and inhibits SCAP through a novel protein degradation pathway, SQSTM1-mediated autophagy-independent lysosomal degradation (SMAILD) pathway. Although the mechanism of SQSTM1-mediated lysosomal transport is still not fully elucidated, this discovery broadens our understanding of molecular glens that degrade target proteins independently of either ubiquitin-proteasome or autophagy-mediated lysosomal degradation pathways.
ATTEC targeting lipid droplets
Lipid droplets originate from the endoplasmic reticulum and are ubiquitous cellular lipid storage structures in which the lipid core is covered by a phospholipid monolayer decorated with proteins. Lipid droplets biosynthesis/degradation and their interactions with other organelles, such as mitochondria, regulate cellular lipid metabolism and energy balance. When energy is in excess, lipid droplets store excess energy in the form of fat, mainly triglycerides and cholesteryl esters. When the body needs energy, lipid droplets release stored lipids for the normal life activities of cells. However, excessive lipid droplets can lead to the increase of adipose tissue and its massive deposition in non-fatty tissues (such as the liver, heart, and muscle), leading to neurodegenerative diseases and metabolic syndrome, such as obesity, diabetes, fatty liver, atherosclerosis, cardiovascular disease, etc.
Therefore, effective inhibition or regulation of the level of lipid droplets is a means of treating obesity and its related metabolic diseases. The Fu group selected GW5074 (GW) and 5, 7-dihydroxy-4-phenylcoumarin (DP) as the "target heads" for LC3 binding and designed two ATTECs (LD·ATTEC1 (C1) and LD·ATTEC2 (C2)) (Fig 5). They confirmed their effectiveness in targeting protein degradation by subsequent cell experiments.
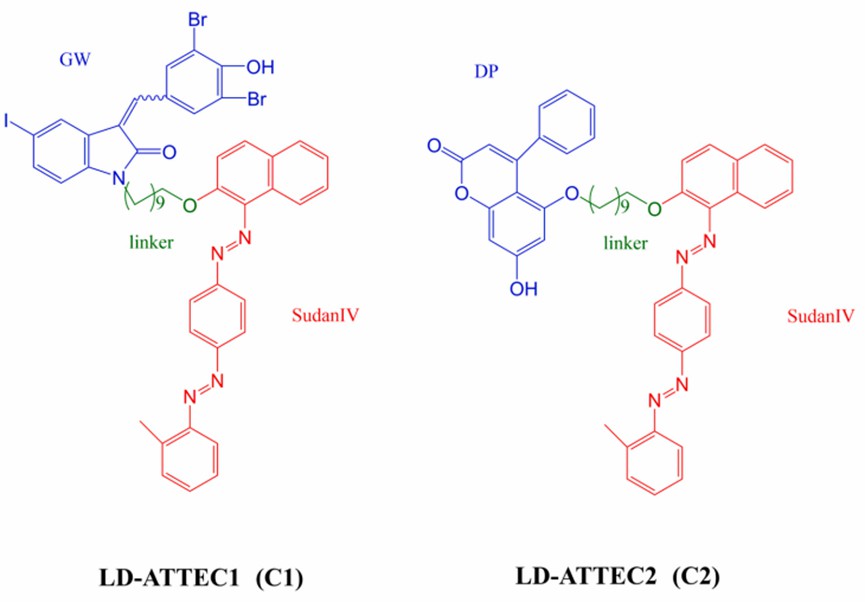 Fig 5. The structure of ATTEC targeting lipid droplets
Fig 5. The structure of ATTEC targeting lipid droplets
LD·ATTECs effectively attenuated the metabolic disease phenotype in vivo, further demonstrating the feasibility of LD·ATTECs treatment. Currently, the team has synthesized 24 LD·ATTECs with different linker and target heads and tested their efficacy.
Summary of PROTACs in Metabolic Syndrome
Nowadays, targeted protein degradation technology is still a hot topic in the field of drug research and development. Compared with the increasingly competitive field of cancer treatment, the treatment of metabolic diseases still has great development potential and challenges. Different from the inhibition of conventional drugs, targeted protein degradation technology directly regulates protein levels by eliminating pathogenic proteins. Efficient removal of misfolded proteins, intracellular and extracellular aggregated proteins, functionally defective organelles, and other toxic macromolecules can be achieved by different degradation strategies. This also means that the range of alternative targets is fully extended. Previous studies have identified many pathogenic targets in metabolic syndrome, but traditional small molecule inhibitors cannot effectively bind such targets with no obvious binding pocket. With the introduction of targeted protein degradation technology and the confirmation of more metabolism-related targets, we believe that protein degradation technology represented by PROTAC will bring new vitality to the treatment research of metabolic diseases.
