Introduction to Enzyme Function and PROTAC
Classical enzymes usually consist of enzyme domains and non-enzyme domains. The functional domain of an enzyme is the region with a specific spatial structure where the enzyme performs its catalytic function. Specifically, the enzyme active site contains a binding region and a catalytic region, which participate in the binding of substrates and catalyze specific chemical reactions of substrates, respectively. The non-enzyme functional region regulates substrate protein activity mainly through protein-protein interaction (PPI), which is independent of catalytic function and mediates the interaction between substrate and different protein components in signaling pathways, such as allosteric regulation and scaffold protein function.
Many studies have disclosed that the non-enzymatic functional regions of various target proteins are involved in the regulation of cell division, differentiation, RNA metabolism, DNA repair, and genome stability. In addition, the functions of these non-enzymes are closely related to various human diseases (e.g., cancer and cardiovascular disease) and play a key role in regulating cell signaling and determining cell fate. However, traditional small molecule inhibitors usually directly target the catalytic functional domain of the enzyme and exert therapeutic effects by inhibiting the enzymatic function of the target protein. As a consequence, these inhibitors usually fail to block the non-enzymatic function of the target protein and face problems of low selectivity, poor specificity, limited clinical efficacy, and drug resistance.
Targeted protein degradation (TPD) is considered a promising and attractive therapeutic strategy. PROTAC is usually composed of three parts: a target protein ligand, an E3 ubiquitin ligand, and a linker. PROTAC recruits E3 ubiquitin ligases to promote ubiquitination and degradation of target proteins through the ubiquitin-proteasome system (UPS).
Unlike traditional drug development strategies that directly inhibit target proteins, PROTAC works by modulating the host protein degradation system. In addition, PROTAC can induce degradation of the entire target protein and block both enzymatic and non-enzymatic functions of the target protein. Therefore, blocking the non-enzymatic function of the target protein through the PROTAC strategy may be a new and promising therapeutic strategy to solve the problems faced by traditional small molecule inhibitors.
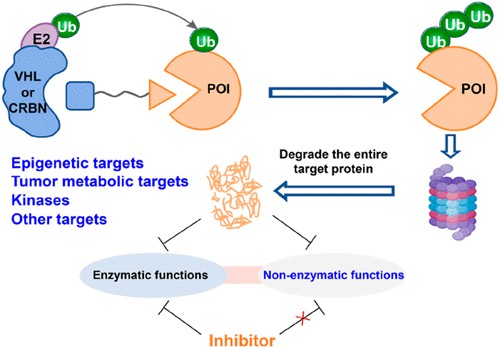 Fig 1. PROTAC technology targeting enzyme structure1
Fig 1. PROTAC technology targeting enzyme structure1
1. PROTACs that block epigenetic target non-enzymatic function
-PROTAC targeting EZH2
Epigenetics is the study of genetic modification of gene expression without changing the nucleotide sequence of genes. Abnormal epigenetic modification widely exists in the development of a tumor and is a hot topic in the development of antitumor drugs.
PRC2 has histone methyltransferase activity and is an important target for epigenetic cancer therapy. PRC2 complex consists of EZH2, EED, SUZ12, and RbAp46/48. Overactivation of the PRC2 complex induces malignancy by silencing tumor suppressor genes. EZH2 is a core and multifunctional catalytic subunit of PRC2 that is overexpressed in a variety of cancer types, such as lung, bladder, and breast cancers. EZH2 inhibits the expression of tumor suppressor genes by methylating lysine 27 at histone H3.
In recent years, significant progress has been made through the development of inhibitors that directly or indirectly target EZH2. Increasing evidence revealed that the carcinogenic function of EZH2 is not fully dependent on its enzyme activity. In addition to catalyzing H3K27me and mediating gene silencing associated with various cellular processes, EZH2 also mediates the activation of genes in various cancers that are not related to the enzymatic function of EZH2/PRC2. However, currently reported EZH2 inhibitors downregulate H3K27me3 levels only by targeting their histone methyltransferase activity. Due to inadequate inhibition of EZH2's carcinogenic activity, EZH2 inhibitors have limited clinical efficacy and are only effective against certain cancers. Therefore, the targeted protein degradation strategy PROTAC provides a new opportunity to completely block the carcinogenic activity of EZH2.
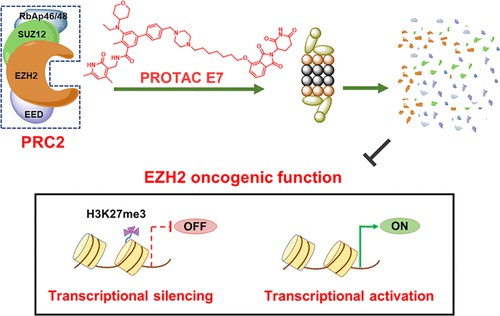 Fig 2. The structure of PROTAC 1 targeting EZH22
Fig 2. The structure of PROTAC 1 targeting EZH22
-PROTAC targeting HDAC6
Histone deacetylase 6 (HDAC6), a microtubule-associated member of the HDAC family, is located primarily in the cytoplasm and is involved in regulating the degradation, cell morphology, and migration of misfolded proteins. Abnormal regulation of HDAC6 is closely related to cancer, neurodegenerative diseases, autoimmune diseases, and many other diseases. Selective inhibitors of HDAC6 are known to block enzyme function by binding to the C-terminal catalytic domain of HDAC6. However, due to the presence of multiple domains in HDAC6, such as the C-terminal ubiquitin-binding domain (UBD), N-terminal catalytic domain, and zinc-finger ubiquitin binding domain (ZNF-UBP), as many as 45 HDAC6 inhibitors currently fail to target these functional domains. Therefore, a new HDAC6 development strategy is urgently needed.
Several PROTACs targeting HDAC6 have been reported. Compound 9 is a VHL ligand-based PROTAC, and compounds 12, 13, and 14 are CRBN ligand-based PROTACs (compounds 13 and 14 are negative controls). These PROTACs show significant HDAC6 degradation and inhibition of tumor cell growth. These depressants block both enzymatic and non-enzymatic functions of HDAC6 by inducing the degradation of the whole target protein.
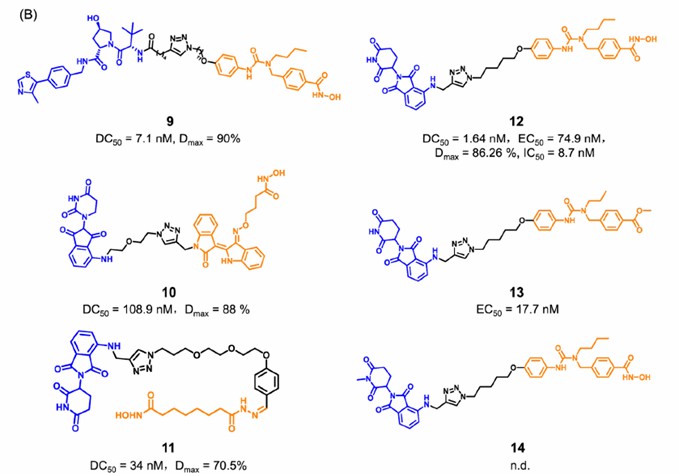 Fig 3. The structure of PROTACs targeting HDAC61
Fig 3. The structure of PROTACs targeting HDAC61
2. PROTACs that block the non-enzymatic function of tumor metabolic targets
Nicotinamide phosphoribosyltransferase (NAMPT) is a key enzyme in the biosynthesis of nicotinamide adenine dinucleotide (NAD), which plays a vital role in tumor metabolism and inflammation. In addition to its role in tumor cell proliferation and differentiation, NAMPT also affects the immune microenvironment due to its cytokine-like effects.
NAMPT is a pleiotropic protein that exists in two distinct forms: intracellular NAMPT (iNAMPT) and extracellular NAMPT (eNAMPT). It has been shown that NAMPT inhibitors only block enzyme function and do not regulate non-enzyme function through eNAMPT. Only inhibiting the enzyme function of NAMPT is not enough to completely inhibit the carcinogenic function of NAMPT. Clinical trials of two NAMPT inhibitors (FK866 and CHS-828) are discontinued due to limited antitumor efficacy and dose-dependent toxicity (e.g., thrombocytopenia and gastrointestinal side effects). Therefore, new strategies are needed to interfere with the non-enzymatic function of NAMPT.
The first PROTAC 15 has been reported to degrade NAMPT and reduce secretion by eNAMPT. Compound 15 directly degrades iNAMPT through the UPS pathway, thereby reducing the secretion of eNAMPT and promoting anti-tumor immunity, thus blocking both enzymatic and non-enzymatic functions of NAMPT. Compound 15 also shows a good inhibitory effect in tumor mouse models.
In addition, compound 15 activated the immune response with lower cytotoxicity and better pharmacokinetic properties. The development of PROTAC 15 has enabled us to better understand the role of NAMPT's non-enzymatic function in reconstructing immunosuppressive tumor microenvironments, thus facilitating the development of NAMPT-targeted tumor immunotherapy approaches.
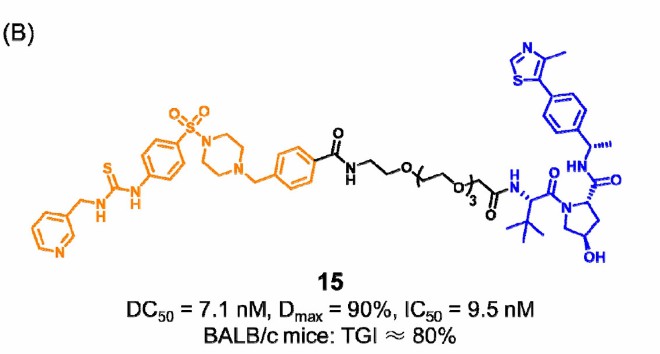 Fig 4. The structure of PROTACs targeting NAMPT1
Fig 4. The structure of PROTACs targeting NAMPT1
3. PROTACs that block the non-enzymatic function of kinases
-PROTAC targeting the mitotic kinase AURORA-A
The mitotic kinase AURORA-A matters in the phosphorylation of various proteins during mitosis, and its catalytic activity is critical for the entire cell cycle. AURORA-A is considered an important target for anticancer drug discovery. However, the development of AURORA-A kinase inhibitors has stalled due to low clinical response rates. It has been shown that the non-enzymatic functional region of AURORA-A can bind to the proto-oncogene proteins of the MYC family, preventing N-MYC and C-MYC from being degraded by the proteasome. Moreover, AURORA-A kinase inhibitors are not sufficient to eliminate the carcinogenic activity of AURORA-A.
To explore the non-enzymatic function of AURORA-A kinase, Wolf's research group developed PROTAC JB170 by binding alisertib, a clinical inhibitor of AURORA-A, to the E3 ligand of CRBN. JB170 was found to induce rapid, effective, and highly specific degradation of AURORA-A. The activity of AURORA-A was thought to be mainly in the G2/M phase of the cell cycle, and its function in the S phase may be independent of its enzyme activity. The researchers found that JB170 induced a strong S phase arrest, but had no significant effect on cell aggregation in the G2/M phase. In addition, researchers demonstrated that the JB170 block S phase is mainly caused by the non-enzymatic function of AURORA-A during DNA replication.
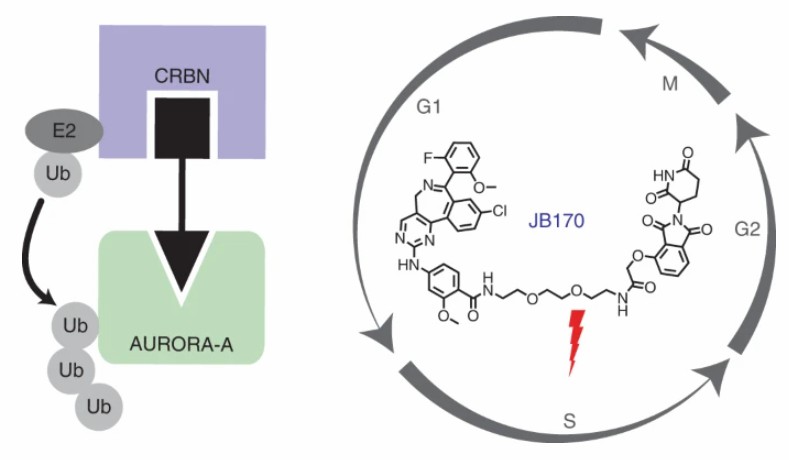 Fig 5. The structure of JB1703
Fig 5. The structure of JB1703
-PROTAC targeting FAK
FAK is a cytoplasmic protein tyrosine kinase that mediates growth factor receptor and integrin-related signal transduction. FAK is mainly composed of three domains: the N-terminal FERM domain, the central kinase domain, and the C-terminal focal adhesion targeting (FAT) region. Although conventional FAK inhibitors only act on protein kinase domains to block enzyme function, the non-enzymatic function of FAK is also critical in the development and progression of cancer and cannot be blocked by reported FAK inhibitors. Therefore, inhibition of both kinase-dependent enzyme function and kinase-independent scaffold function of FAK is a new research direction for antitumor drug development.
4. PROTACs that target non-classical functional proteins
-PROTAC targeting FKBP12
Pulmonary arterial hypertension (PAH) is the most common and serious complication of congenital heart disease, and also the main problem of cardiovascular disease prevention and treatment. Recent studies have found that iron plays a crucial role in the occurrence and development of PAHs. Bone morphogenetic protein (BMP) is one of the key proteins in PAH and participates in the expression of hepcidin, a key protein regulating iron metabolism balance in the human body.
FK506 binding protein 12 (FKBP12) binds to BMP type I receptors and subsequently inhibits hepcidin. This non-classical function of FKBP12 is similar to the non-enzymatic function in that it regulates its interaction with different components of the cell signaling pathway in a catalyst-independent manner. Currently reported immunosuppressants rapamycin and FK506 block FKBP12 binding to BMP type I receptors, thereby increasing hepcidin. However, in clinical use, FKBP12 inhibitors exhibit immunosuppressive side effects.
-PROTAC targeting USP7
As an important cancer suppressor, p53 is one of the most frequently mutated genes in cancer. P53 mutates in more than 50% of human cancers. Studies have found that mutated p53 not only loses the regulation of the normal biological function of cells but also inhibits the function of wild-type p53 protein, thus leading to cell cancer. Therefore, the development of new drugs to treat cancers with p53 mutations remains an urgent problem. However, p53 mutations are relatively random, making it difficult to develop targeted drugs that directly target p53 mutants.
Ubiquitin-specific protease 7 (USP7) regulates p53 levels by deubiquitinating and stabilizing MDM2. Recently, a Chinese research group from Shanghai Pharmaceutical Institute designed and synthesized the first-generation small-molecule depressant U7D-1, which can effectively and selectively degrade USP7 (DC50=33 nM). Compound U7D-1 showed a stronger inhibitory activity against the growth of p53 wild-type cancer cells than the USP7 inhibitor, especially in p53 mutant cancer cells, while USP7 inhibitor 27 showed weak activity. Further studies of the mechanism of action suggest that U7D-1 may induce the degradation of USP7 by regulating the non-enzymatic functional region of USP7 (apoptotic and E2F pathways), thus exerting its antitumor activity against p53 mutant cancer cells.
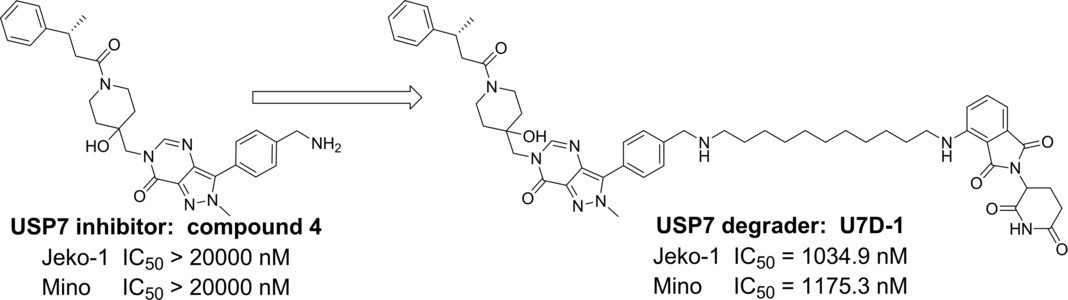 Fig 6. The structure of PROTAC U7D-1 targeting USP74
Fig 6. The structure of PROTAC U7D-1 targeting USP74
Summary
In recent years, the non-enzymatic function of target proteins has attracted increasing attention. PROTAC can block both enzymatic and non-enzymatic functions of the target protein at the same time and occupy the enzyme binding pocket to induce the degradation of the whole protein for a short time.
Present studies reveal that PROTAC has advantages in studying the non-enzymatic function of the target protein and plays an effective therapeutic role. Compared with small molecule inhibitors, PROTAC has higher target selectivity, stronger efficacy, lower risk of resistance, and prolonged action.
Therefore, PROTAC-mediated inhibition of enzymatic and non-enzymatic functions of drug targets can lead to significant improvement of drug activity, providing a new therapeutic strategy and a basis for the study of non-enzymatic functions of target proteins and the mechanism of related diseases. Although the research on the non-enzymatic function of PROTAC is still in its infancy and more studies are needed to discover more strategies to regulate the non-enzymatic function of target proteins, it is believed that using PROTAC to block the non-enzymatic function of proteins may be a promising new direction for drug development.
References
- Sun, D., et al. Blocking Non-enzymatic Functions by PROTAC-Mediated Targeted Protein Degradation, J. Med. Chem. 2022, 65, 21, 14276–14288.
- Liu, Z., et al. Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2, J. Med. Chem. 2021, 64, 5, 2829–2848.
- Adhikari, B., et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase, Nature Chemical Biology. 2020, 16, 1179–1188.
- Pei, Y., et al. Discovery of a Potent and Selective Degrader for USP7, Angew. Chem., Int. Ed. Engl. 2022, 134 (33), e202204395.
