PROTAC degraders have emerged as a groundbreaking modality in targeted protein degradation, offering a novel approach to eliminating disease-related proteins rather than simply inhibiting them. This article provides a comprehensive summary of PROTAC compounds currently in clinical trials, highlighting key molecules, therapeutic targets, and clinical outcomes. Whether you're tracking ARV-110, CFT7455, or other leading candidates, this overview delivers up-to-date insights into the clinical landscape of PROTAC-based therapeutics.
PROTACs has emerged as an attractive and innovative strategy for modulating protein of interest (POI) via degradation. PROTACs are hetero bifunctional molecules which consist of a POI ligand and an E3 ubiquitin ligase (E3) recruiting ligand joined by an optimal linker. The mechanism begins when PROTACs facilitate a ternary complex formation between POI and E3. The ubiquitination of the POI then takes place once the ubiquitination machinery is brought within proximity, and lastly the ubiquitinated POI was identified and degraded by the 26S proteasome (Ubiquitin–proteasome system (UPS) in eukaryotic cells). PROTACs, in partnership with the UPS system, provides the possibility for regulating protein levels. In other words, PROTACs is a chemical knockdown strategy.
What Are PROTACs?
PROTACs are hetero-bifunctional small-molecule degraders that enlist the cellular UPS to degrade a POI rather than merely block its activity. PROTACs are trisected into three pharmacologically independent modules: an inducer moiety that binds the POI, a binder that engages an E3 ubiquitin ligase, and a chemical linker that juxtaposes these two recognition domains in three-dimensional space. Since the molecular-recognition elements are modular, in principle any POI for which a small-molecule or peptide binder exists, including enzymes, scaffolds and transcription factors, can be targeted for degradation. PROTACs reported in 2001 utilized a peptidic E3 ligase ligand to prove the proof-of-concept for inducing the degradation of methionine aminopeptidase-2 (MetAP-2). Medicinal-chemistry efforts in subsequent years have focused on replacing these peptide motifs with drug-like small molecules targeting E3 ligases such as VHL, CRBN, MDM2 and IAPs in order to improve cell permeability, metabolic stability, and synthetic tractability. PROTACs as a result, have carved out a unique role in the modern pharmacopoeia: they extend the druggable proteome to the ~85 % of human proteins that were previously deemed "undruggable" due to a lack of catalytic pockets for occupancy-driven inhibitors. PROTACs are being explored for therapeutic utility in neurodegeneration, autoimmune disorders, viral infections, and cardiovascular pathologies and more than thirty candidates have entered clinical trials since 2019. PROTACs' catalytic mode of action, event-driven pharmacology, and circumvention of classical resistance mechanisms makes them a distinct therapeutic modality that is well-poised to not only complement but also in some cases supplant traditional small-molecule inhibitors.
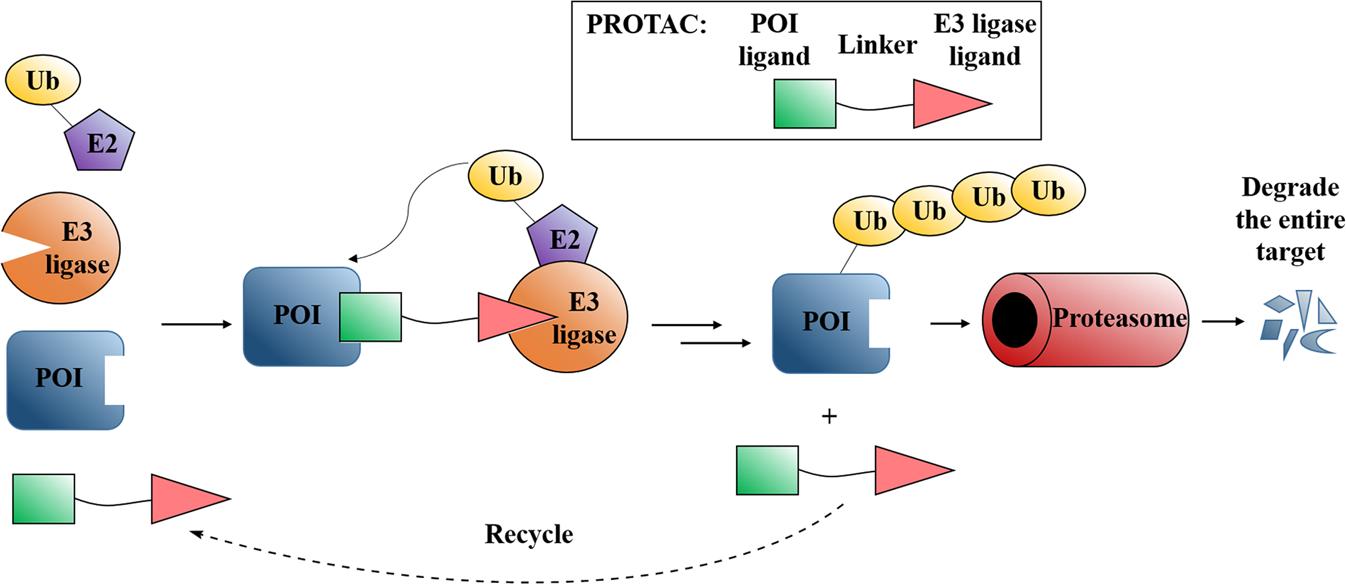 Fig. 1 Mode of action of PROTACs.1,2
Fig. 1 Mode of action of PROTACs.1,2
How Do PROTACs Work in Targeted Protein Degradation?
Mechanistically, PROTACs act as "molecular glues" that enforce proximity between a POI and an E3 ubiquitin ligase, co-opting endogenous UPS components for selective proteolysis. PROTACs bind their cognate POI and an E3 ligase in parallel upon cellular uptake, and a ternary complex is formed via cooperative protein–protein interactions. In the ternary complex, the E3 ligase facilitates the covalent linkage of multiple ubiquitin moieties to the surface lysine residues of the POI, and the resulting poly-ubiquitin signal is recognized by the 26S proteasome. The tagged POI is unfolded and the protein is cleaved into short peptides, and the unoccupied PROTAC is released to start a new catalytic cycle. As PROTACs do not need to bind with high affinity to an active site to be effective, they are applicable to target transcription factors, scaffolding proteins, and mutant oncoproteins that otherwise have shallow or transient binding pockets. In addition to enabling the use of sub-stoichiometric dosing, PROTACs' catalytic efficiency also has key pharmacological implications: they can maintain long-lasting target knockdown at lower systemic exposures, thereby lowering the dosing frequency and potential for off-target liabilities relative to classical inhibitors. Because PROTACs ablate all functional domains of a POI, including scaffolding surfaces and protein–protein interaction domains, they can bypass resistance mechanisms stemming from point mutations that only impair active-site binding but have little to no effect on ligand-induced degradation. Structural studies of POI–PROTAC–E3 complexes have also revealed their dynamic conformational landscapes, guiding rational linker optimization and next-generation degrader design. Overall, these and other mechanistic insights continue to drive the maturation of PROTACs as a versatile and powerful platform for targeted protein degradation.
Key PROTACs in Clinical Trials
PROTACs are usually in the 700–1000 Da size range. PROTACs are more challenging to administer orally due to their larger size, but they act like ordinary small molecules once they are in circulation. The molecules in this size range act like one would expect from little molecules when they are intravenously, intraperitoneally, or subcutaneously delivered: exposures are inversely related to the dose, the molecules are well-distributed, and little liver clearance is observed for optimized chemicals. Most importantly, in vivo target proteins are extensively degraded, as demonstrated by many studies, during preclinical development. In tumor tissue, for example, >90% breakdown of Brd4 has been shown as a result of subcutaneous administration of a Brd4 PROTAC. For these reasons, most challenges associated with making PROTACs available are addressed with traditional medicinal chemistry techniques. Owing to PROTACs' extraordinary catalytic efficiency, target degradation and the consequent efficacy in preclinical models can often be demonstrated at plasma concentrations as low as 100–200 nanomolar. As a result, the distribution strategy is influenced by the choice of target protein and disease indication to a large extent. Administering a PROTAC intravenously is the quickest way to the clinic if the effect can be reached with intermittent dosing, such as by killing tumor cells or eliminating a toxic, aggregated protein. Intravenous administration also avoids the gastrointestinal absorption phase required for oral drugs, and a wide range of well-characterized prescription formulations are applicable to PROTACs. A low dose is also typically expected to be sufficient for therapeutic efficacy due to PROTACs' unusually high potency.
Current Leading PROTACs Undergoing Clinical Evaluation
The design and development of a PROTAC is a lengthy process and not all biological targets may be eligible for protein degradation via this mechanism. Thus, it would be quite useful to get an early confirmation of the POI goals. It was demonstrated that a number of therapeutic kinase targets were susceptible to PROTAC-induced degradation. Similar studies might be helpful for other protein families before starting PROTAC development. In addition, techniques for protein labeling like HaloTag (HT) and His-tagged POI can be employed to determine the degradability of a target for target validation. The majority of target proteins that have been developed so far for PROTACs are still part of the druggable proteome. It might be possible to develop molecules that can modulate non-traditional pharmacological targets which are difficult to modify with the use of PROTAC technology. PROTAC technology only necessitates binders that transiently mediate ternary compound formation, allowing for the inclusion of low affinity POI ligands. 15 PROTACs will have entered clinical trials by the end of 2021.
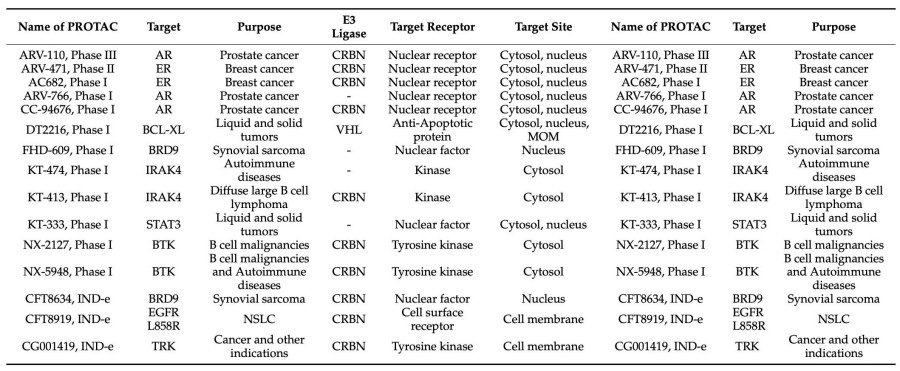 Fig. 2 PROTACs in Clinical Trials by the End of 2021.3,4
Fig. 2 PROTACs in Clinical Trials by the End of 2021.3,4
Successes and Setbacks in Clinical Trials
ARV-471 became the first PROTAC to enter two Phase III studies in parallel (VERITAC-2 and VERITAC-3) assessing monotherapy and combination with palbociclib in endocrine-refractory breast cancer. ARV-110 recently completed a Phase II dose-expansion in mCRPC patients harbouring AR ligand-binding-domain mutations, and reported a ≥50 % PSA decline in ~40 % of the biomarker-selected subgroup. DT2216 remains the only VHL-recruiting degrader in the clinic, with its platelet-sparing profile explained by low VHL expression in megakaryocytes. Collectively, these programmes exemplify the clinical maturation of TPD, with CRBN-based degraders dominating the pipeline (>70 %), followed by a more limited VHL-based cluster focusing on anti-apoptotic proteins.
Challenges in Clinical Development of PROTACs
Pharmacokinetics and Toxicity Issues
The clinical advancement of PROTACs is hampered by a suite of PK and toxicological liabilities that are largely unique from traditional small molecules. First, the size (~800–1,200 Da) and high topological polar surface area of most PROTACs breach Lipinski's rule-of-five and result in poor aqueous solubility, limited oral bioavailability and erratic absorption. ARV-110 and ARV-471 displayed non-linear exposure above 200 mg in early-phase trials because of solubility-limited absorption; amorphous solid dispersions and lipid-based self-emulsifying drug delivery systems have since been developed to enable consistent systemic delivery. Second, PROTACs are rapidly metabolised by first-pass hepatic CYP3A4 and UGT1A1; linker deuteration or the inclusion of metabolically robust heteroaromatic rings (e.g., triazoles) has been shown to prolong from 1–2 h to >8 h in rodents. Third, the catalytic, event-driven mechanism of PROTACs results in a unique "hook effect": supra-optimal doses favor binary (POI–PROTAC or E3–PROTAC) rather than ternary complexes, counterintuitively reducing degradation efficiency and complicating traditional MTD trial design. Fourth, there is the potential for off-target toxicity. CRBN-recruiting degraders can non-selectively induce degradation of the transcription factors IKZF1/3 and GSPT1, leading to immunosuppression and thalidomide-like thrombo-embolic events. VHL-based PROTACs targeting BCL-XL (e.g., DT2216) have been shown to cause transient thrombocytopenia in certain disease contexts when VHL is highly expressed in megakaryocytes. Finally, tissue-selective delivery is complicated by the cell-contextual availability of E3 ligases; CRBN, VHL and MDM2 expression can vary by >50-fold across tumor histologies and normal tissues, requiring patient stratification based on ligase immunohistochemistry or transcriptomics. PROTACs encapsulated in tumor-targeted nanoparticles, antibody–PROTAC conjugates or light-activated "opto-PROTACs" are being developed to limit degradation to cancerous cells while preserving normal tissues.
Target Identification and Selectivity
Key bottlenecks that have limited the clinical reach of PROTACs thus far are selecting the right target and achieving exquisite selectivity. Despite initial promise as a strategy to target "undruggable" proteins, only a few degraders have been able to engage such targets in vivo to date; the majority of clinical stage PROTACs are still directed towards well-established kinases, nuclear hormone receptors or epigenetic "reader" proteins. The bottleneck is the discovery of high quality ligands with (i) an empirically measurable affinity for the POI, (ii) an accessible lysine residue for ubiquitination, and (iii) low conformational restriction within the emerging ternary complex. For protein–protein interaction (PPI) surfaces, which tend to be flat and hydrophobic, fragment-based screening approaches combined to structure-guided linker extension have led to only ~50 validated POI–PROTAC pairs over the past 10 years. In addition, having a POI ligand available is no guarantee of selectivity. PROTACs can be differentially selective even for closely related paralogues. For example, BET degraders have been shown to be more efficient at depleting BRD4 vs. BRD2/3 due to minor differences in accessibility of surface lysines. In other cases, non-intended "neo-substrate" degradation may emerge if the E3 ligase ligand (e.g., thalidomide analogues) crosslinks non-cognate proteins in a "molecular glue" manner, expanding the toxicology footprint. To avoid such pitfalls, chemoproteomic methods such as thermal proteome profiling (TPP) and isotopic tandem-mass-tag (TMT) pulldowns are being employed as early as possible in the optimisation cycle to chart the global degradome of each PROTAC candidate. Machine-learning algorithms trained on >2,000 ternary complex structures are also being leveraged to identify off-target liabilities and prioritise linker chemistries that enhance cooperativity while limiting entropic costs. Finally, diversifying the E3 ligases beyond the well-trodden CRBN/VHL/MDM2/IAP axis is also a key priority for tissue-restricted targeting. Several recent academic consortia have taken the first steps towards establishing systematic ligand discovery programmes for TRIM21, DCAF15, RNF43 and FEM1B, with initial probes showing promising selectivity in neuronal and haematopoietic lineages. Together, these efforts are starting to shift the target-selection paradigm from "any ligand will do" to a more rigorously validated, context-specific degrader design approach.
Future Outlook for PROTACs in Medicine
Potential Applications in Cancer and Neurological Diseases
The therapeutic potential of PROTACs is being realized at an accelerating rate, largely because of the unique ability to eradicate, rather than transiently suppress, pathogenic proteins. In oncology, the field's most mature focus area, PROTACs are being developed against three main target categories: historically "undruggable" transcription factors (e.g., MYC, STAT3 and β-catenin) for which occupancy-based inhibitors have not been successful; kinases that develop allosteric or gate-keeper mutations (e.g., BTK, EGFR) that are resistant to ATP-competitive drugs; and epigenetic "reader" proteins (BRD2/3/4, EZH2) whose scaffolding function drives oncogenic transcription. In pre-clinical models, a single i.v. dose of a CRBN-recruiting BET-PROTAC can achieve >90 % tumour regression in MYC-driven xenografts along with suppression of downstream MYC signatures, which has not been possible with the BET inhibitor JQ1 at tolerated exposures. In solid tumours, PROTACs are being developed to increase tumour penetration through nano-formulations, and conjugation to antibodies that target over-expressed surface antigens (e.g., HER2-PROTAC conjugates for HER2 breast cancer) to spatially confine degradation and reduce systemic toxicity. In the neurosciences, efforts are transitioning from exploratory proof-of-concept to bona fide therapeutic programmes. The most advanced focus is AD, with recent reports describing tau-directed PROTACs that cross the BBB through intranasal or intracerebroventricular delivery and suppress pathological tau by >95 % in hippocampal neurons of 3×Tg-AD mice within 24 h of dosing. A second-generation molecule, QC-01-175, discriminates mutant tau from wild-type isoform via a Keap1-Cul3 E3 ligase, thereby sparing the physiological functions of tau.
How PROTAC Technology Could Revolutionize Drug Discovery?
PROTACs may offer a fundamental shift to the pharmaceutical R&D paradigm, where the notion of "druggability" is no longer a binary entry criterion but a spectrum. Whereas conventional drug discovery requires high-affinity pockets leading to attrition rates >90 % for proteins with no or shallow catalytic clefts, PROTACs turn this approach on its head. PROTACs only need ligandability (binding with micromolar affinity), since the energetic penalty for degradation is offset by the ubiquitin-proteasome system and not the drug itself. This has already led to an estimated 20-fold increase in targetable proteome and now spans so-called transcription factors (STAT3, MYC), scaffolding proteins (β-catenin), and aggregation-prone proteins (tau, huntingtin) that had previously been classified as "undruggable". The catalytic, event-driven mode of action also provides several advantages for translation. First, sub-stoichiometric dosing can reduce the systemic exposure and associated liabilities. For example, ARV-471 induces maximal degradation of estrogen-receptor at plasma concentrations that are 100-fold lower than the IC₉₀ of the PROTAC's comparator fulvestrant. Second, degradation obviates adaptive resistance mechanisms such as point mutations in the kinase active site. BTK-resistant CLL cells are still sensitive to CRBN-recruiting BTK-PROTACs because the C481S mutation does not interfere with ligand-mediated ubiquitination. Third, PROTACs can be "re-programmed" modularly by exchanging either the POI ligand or the E3-recruiting motif, allowing for rapid repurposing of an asset once a validated pharmacophore pair is identified. This plug-and-play architecture has enabled the design of dual-degrader constructs that simultaneously degrade BRD4 and CDK4/6, and can induce synthetic lethality in RB-deficient breast tumors with a single molecule.
How Our Products and Services Accelerate PROTAC Development?
As clinical trials for PROTAC degraders continue to expand globally, access to reliable, high-purity reagents and expert scientific support has never been more critical. At BOC Sciences, we empower biotech and pharmaceutical innovators with a full range of PROTAC products and services designed to support discovery, optimization, and translational research. Whether you're validating a degrader target or scaling up for preclinical development, we provide the tools and expertise to keep your program moving forward-efficiently and confidently.
Our High-Quality PROTAC Chemicals and Reagents
We offer a broad portfolio of research-grade and GMP-ready PROTAC building blocks and reference compounds to support every stage of your workflow:
- E3 ligase ligands including CRBN, VHL, IAP, MDM2, and more.
- Warhead molecules targeting a wide range of proteins (kinases, epigenetic targets, transcription factors, etc.).
- Optimized linkers and linker libraries for rapid structure–activity exploration.
- Ready-to-use PROTAC reference compounds for benchmarking and method development.
- Purity-verified materials with full documentation (COAs, MS/NMR spectra) to meet regulatory standards.
All our chemicals undergo stringent quality control to ensure reproducibility and consistency in your PROTAC development pipeline.
Custom PROTAC Design and Consultation Services
Looking to design a novel PROTAC molecule? Our end-to-end custom synthesis and consultation services give you the flexibility to bring your concepts to life with scientific precision and speed. Services include:
- Target-specific PROTAC design, integrating your warhead of choice with the most suitable E3 recruiter.
- Molecular modeling and linker optimization using AI-enhanced tools and 3D structural analysis.
- Custom synthesis of PROTACs, intermediates, and analogs, with scalable batch options.
- In vitro degradation assay support to validate efficacy and selectivity in relevant cellular models.
- Project-based consultation from experienced medicinal chemists and protein degradation specialists.
Whether you're designing a PROTAC for a novel target or optimizing an existing lead, our team acts as your extension—delivering scientific solutions aligned with your R&D goals.
Looking to accelerate your own PROTAC research? We offer a comprehensive portfolio of high-purity PROTAC compounds, linkers, E3 ligase ligands, and custom synthesis services. Partner with us to gain a competitive edge in your clinical development pipeline. Explore our PROTAC product catalog today.
References
- Image retrieved from Figure 1 " Mode of action of PROTACs," Sun X.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Sun X, Gao H, Yang Y, et al. PROTACs: great opportunities for academia and industry[J]. Signal transduction and targeted therapy, 2019, 4(1): 64. https://doi.org/10.1038/s41392-019-0101-6.
- Image retrieved from Table 2 " PROTACs in Clinical Trials by the End of 2021," Sincere N I.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Sincere N I, Anand K, Ashique S, et al. PROTACs: emerging targeted protein degradation approaches for advanced druggable strategies[J]. Molecules, 2023, 28(10): 4014. https://doi.org/10.3390/molecules28104014.3)
- Kubryń N, Fijałkowski Ł, Nowaczyk J, et al. PROTAC Technology as a New Tool for Modern Pharmacotherapy[J]. Molecules, 2025, 30(10): 2123. https://doi.org/10.3390/molecules30102123.
- Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy[J]. Journal of hematology & oncology, 2020, 13(1): 50. https://doi.org/10.1186/s13045-020-00885-3.
- Han X, Sun Y. PROTACs: A novel strategy for cancer drug discovery and development[J]. MedComm, 2023, 4(3): e290. https://doi.org/10.1002/mco2.290.
