Far from being an inert tether, the linker is the dynamic backbone of a PROTAC: its length determines the radius of collision between E3 ligase and target protein, its flexibility modulates the likelihood that such an encounter will adopt a ubiquitination-competent geometry, and its polarity governs whether the trimer survives long enough in aqueous cytosol to be productive. Because these three variables are inter-dependent (short, rigid segments may crystallize into stable complexes yet fail to reach membrane interiors, while floppy, hydrophilic chains dissolve readily but sample too many unproductive conformations), linker optimization has matured into a systems-level exercise coupling computational conformer libraries to micro-scale synthesis and live-cell degradation read-outs.
Introduction — Why Linkers Matter?
In traditional medicinal chemistry, affinity and exposure are treated as attributes of the ligand itself; in the PROTAC space, they are emergent properties of a three-body problem whose choreography is scripted by the linker. A difference of two methylene units can switch a degrader from potent to inert, not because either pharmacophore has changed, but because the new span either pre-organizes or forbids the trimeric interface required for ubiquitin transfer. As a result, the linker has become the primary tuning dial through which potency, selectivity, and pharmacokinetics are reconciled without touching the already-optimized warheads.
The Linker as the Bridge Between Two Worlds
In principle, the linker language (orthogonal to the recognition languages of the two proteins – one on the E3 surface and one in the target binding groove) serves to mediate between the two in a mutual spatial vocabulary which is amenable to decoding by the proteasome. In practice, as evidenced by crystal structures, a well-designed linker allows for some conformational freedom and thus serves as a conformational funnel, pushing a nascent complex towards a low-energy path to lysine presentation. Flexible spacers such as oligo-glycines or PEG chains serve as entropy sinks and thus bank the cost of rotational freedom early, so that the two binding partners at the ends of the linker are free to rotate and adapt into each other's pockets. Rigid linkers such as piperazines, alkynes, or bicyclic lactams on the other hand minimize conformational space, which comes at the price of increased productive encounter rates at the expense of less tolerance towards motions of the bound proteins. As a general rule, flexible surfaces such as those often found on transcription factors tend to benefit from linker flexibility, while closed and thus more rigid conformations such as those commonly found on kinases may require a higher degree of directionality. In this way, linker composition also influences hydration shell dynamics: the organized water adsorbed on polyethylene glycol moieties can serve to shield electrostatic repulsion between, for example, acidic patches on the ligase and on the target, while aliphatic linkers tend to displace water molecules and can lead to hydrophobic collapse, effectively reducing the linker distance.
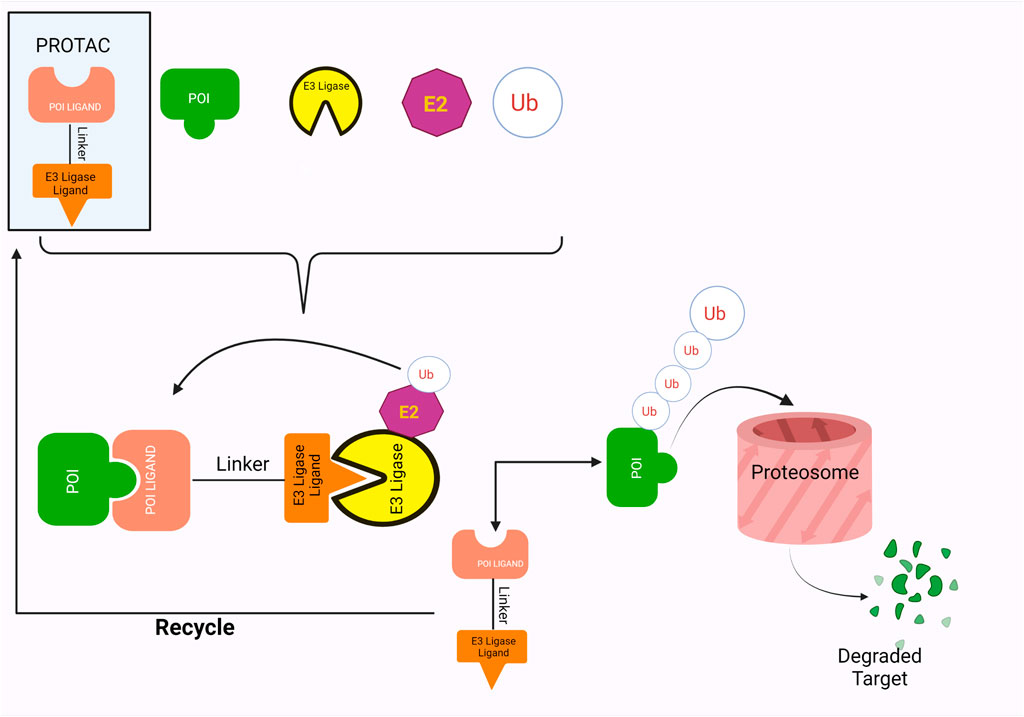 Fig. 1 The PROTACs mode of action, involving ubiquitination and eventual degradation of POI.1,2
Fig. 1 The PROTACs mode of action, involving ubiquitination and eventual degradation of POI.1,2
Its Impact on Target Binding and Complex Stability
In addition to controlling on-rate, the linker also modulates dwell time, the duration of the trimer. This variable has been shown in many cases to have a tighter relationship with degradative potential than with the on-rate of the binary. Linkers that are too short can generate steric strain, which destabilizes the trimer and leads to pulsed ubiquitination events that do not irreversibly commit the substrate to the proteasome. Linkers that are too long permit breathing that dissociates the complex to the point where the E2-loaded ligase can no longer access the lysine ε-amino group, and a previously potent degrader becomes a reversible inhibitor. Additional adjustments can be made through backbone tension; linkers with tertiary amides or ortho-substituted aromatics can form intramolecular H-bonds that sequester polar groups from solvent, reducing effective PSA and increasing cell permeability without affecting the protein-protein contacts. In contrast, an unhindered, centrally placed amide often assumes an extended conformation, increasing the 3D SASA, and impeding transit across the membrane. The same intramolecular H-bonds can in some cases also pre-organize π–π stacking between the linker and aromatic residues in the E3–POI interface, adding enthalpic stabilization to help offset the entropy lost upon rigidification. However, a linker's own metabolic vulnerability must also be considered, as ethers and unsubstituted alkynes are relatively stable while benzylic esters or unprotected phenols are susceptible to oxidative cleavage that would cut the bridge prematurely. As a result, the metric to optimize is not a singular Kd, but rather the integral of productive complex concentration over time, a function almost entirely dictated by linker length, flexibility and polarity.
Physical and Chemical Parameters of Linkers
A PROTAC linker's properties are encoded in three inter-dependent coordinates - the distance the bridge must span, the number of shapes it can sample while spanning, and how freely it dissolves in the aqueous cytosol it must cross. These properties are not additive: a rigid rod's extension by a single phenylene improves the ternary complex half-life but at the same time collapses the membrane permeability, while insertion of a flexible glycol unit may restore solubility at the price of entropic dilution of the active conformer. Successful optimization therefore treats length, flexibility and polarity as a single design surface, to be explored through conformer libraries, micro-scale parallel synthesis and live-cell degradation feedback, rather than by isolated empirical rules.
Length and Conformational Control
Conceptually, the end-to-end distance is probably easier to think of as a probability cloud rather than a physical ruler. All-atom simulations revealed that the most productive trimer geometries aggregate in a narrow window of center-of-mass separations, and that linkers shorter than this are insufficient to fully bridge the distance (compressing the E3 and target into steric conflict), while longer ones permit the ubiquitin-loaded catalytic face to wander out of lysine reach. Thus, conformational control is exercised not only by tuning the atom count but also by pre-programming dihedral preferences. Inserting an alkyne or a 1,4-disubstituted piperazine has the effect of biasing the chain toward an extended rod, while gem-dimethyl or ortho-fluoro substituents favor gauche kinks that shrink the effective span without the need to remove atoms. These small-scale torsional edits can be amplified cooperatively: two consecutive rigid segments are enough to pin the whole linker into a single low-energy arc, while a single flexible junction is sufficient to restore hundreds of transient geometries. The design must also take into account breathing; transcription factors that undergo helix-coil transitions need a linker whose most probable end-to-end distance remains in the productive window even when the target domain expands. The task of length optimization is therefore less about counting atoms than about sculpting the conformer distribution so that the productive sub-population is thermally accessible yet not so dominant that off-target encounters become likely.
Flexibility vs Rigidity: When to Use Each Type
Flexible chains effectively pay the entropy cost up-front (i.e., before binding), so that the terminal ligands are free to sweep through space until they find a complementary pocket. This can be useful if the target surface is itself flexible or if the E3-binding site is shallow and an induced-fit penetration is needed. By contrast, rigid spacers pre-organize the pharmacophores into a geometry that is close to the bound state. The entropic penalty upon complex formation is smaller and the residence time is often extended. The choice between the two is dictated by the mismatch between the unbound and bound states: if, for instance, crystallographic data indicate that the two proteins have to approach one another at an obtuse angle, a semi-rigid linker containing one freely rotatable hinge flanked by two aromatic planes can enforce the directionality without over-constraining the system. However, over-rigidification can backfire by creating a binding site that is too specific: mutations or post-translational modifications on either protein may abolish degradation even while binary affinity is retained. A pragmatic compromise, which has been successful in a number of cases, is to use a chimeric architecture: a short, stiff segment placed close to the E3 ligand that ensures the catalytic face is presented correctly, coupled to a longer, flexible tail attached to the target ligand that accommodates conformational heterogeneity. This hybrid strategy has proved especially useful for degrading kinases that can adopt multiple DFG-in and DFG-out states, where an entirely flexible linker would sample too many inactive poses and an entirely rigid one would fail to recognize the inactive conformation.
Polarity and Solubility Considerations
Polarity affects not only solubility in water but also the linkers' propensity for intramolecular hydrogen bonding that can shield or reveal polar atoms at the protein surface. For this reason, polyethylene glycol chains are favored in part because they attract a shell of ordered water that screens electrostatic repulsion between acidic patches on the ligase and target; the same hydration shell, however, leads to a larger hydrodynamic radius that impedes diffusion across lipid bilayers. In contrast, fully aliphatic chains are more likely to partition into the membrane but risk collapsing into a hydrophobic core that sequesters the terminal ligands within micelle-like structures, thereby reducing the free concentration available for ternary complex formation. A compromise is often a single, ionizable group—morpholine, tertiary amine, or sulfonamide—placed so that its pKa is at least one unit below that of the cytosol and is therefore protonated (and thus soluble) only within acidifying endosomes. Finally, topological polar surface area is a nonlinear property: slight increases can improve oral exposure by preventing self-aggregation, while too much polarity increases the energetic cost of desolvation upon protein binding. As a result, the polarity is tuned relative to flexibility: a rigid, aromatic linker can withstand greater polar surface area because it incurs the desolvation penalty only once, while a flexible chain must repeatedly desolvate each time it samples a new conformer. The last metric is not a single logP value but rather the integral of solubility, membrane permeability and intracellular free fraction—properties that must be co-optimized along with the geometric constraints of the ternary complex.
Structural and Functional Optimization
Optimizing a PROTAC is not necessarily a search for the tightest binder but rather for the most consistent matchmaker: During its function, the linker needs to create a temporary yet recurrent three-body arrangement that positions E3-bound ubiquitin to interact with an adjacent lysine through its particular shape. Because crystal structures represent only a small number of these transient states, modern campaigns often alternate between atomistic modelling that samples thousands of plausible trimer poses and parallel micro-scale synthesis that transforms the top-ranked geometries into testable molecules. The resulting structure–activity landscape is therefore co-authored by silicon and glassware—computational ensembles that suggest which vectors are worth pursuing, and iterative linker libraries that confirm which distances, angles, and flexibilities actually translate into measurable degradation.
Modeling Ternary Complex Formation
In the latest simulations, the linker is no longer modelled as a flexible string. Instead, the calculation is seeded with an ensemble of semi-rigid conformers, whose end-to-end vectors sample a radial grid around the crystallographically observed E3–POI separation. Replica-exchange molecular dynamics allows each segment to "breathe" while the terminal ligands are only loosely restrained in their pockets, producing density maps which emphasize "hot" contact regions where the POI surface repeatedly approaches the catalytic face. These maps are converted into distance probability functions: linkers whose most populated span falls within a narrow window are predicted to stabilize the active geometry, while those sampling bimodal distributions are penalized for entropic dilution. Solvent is explicitly modelled in a hydration shell thick enough to observe water-bridged hydrogen bonds which can glue or repel the partners, an effect especially critical when both proteins display exposed acidic patches. Further refinement introduces backbone flexibility of the POI itself; for kinases that shift between DFG-in and DFG-out states, both conformations are simulated, allowing the algorithm to propose linker lengths that will be productive across the conformational equilibrium. The final output is not a single "best" structure, but a ranked list of geometric hypotheses with predicted strain energies, which are then passed to synthetic chemists as a set of distance–rigidity coordinates rather than as prescriptive atom lists.
Experimental SAR Studies for Linker Selection
Parallel micro-scale synthesis is used to prepare series that vary linker length in single-atom steps, while polarity and rigidity are switched in orthogonal block designs. Every crude reaction mixture is purified by automated solid-phase extraction before being screened in a cell-based degradation assay that reports both target and off-target liabilities in the same well, enabling removal of molecules that, while potently knock-down the target, cause degradation of housekeeping proteins. The resulting data cloud is interrogated with trend-agnostic algorithms that group performance according to emergent physicochemical descriptors—topological polar surface, rotatable-bond fraction, aromatic-to-aliphatic ratio—rather than by a priori categories. Notably, potency often plateaus at a critical linker length beyond which further extension results in no gain but increased metabolic liability; the position of this plateau depends on linker chemistry, with more flexible glycol chains reaching it earlier than semi-rigid alkynyl rods. Rigidity is then introduced gradually: a single alkyne introduced close to the E3 ligand almost invariably improves residence time, but the same alkyne placed nearer to the POI ligand may instead reduce activity if the twist it induces misaligns the lysine trajectory. These findings are then fed back into the modelling cycle, to refine the next virtual library and reduce the chemical space that must be probed in the wet lab. This iterative cycle is repeated until the predicted and observed degradation profiles converge, at which point the linker is considered optimised, not for affinity, but for the integral of productive complex concentration over the PROTAC's cellular lifetime.
Case Studies in Linker Design
Structural comparisons of CRBN- and VHL-recruiting degraders have demonstrated that linker identity cannot be simply copied from one E3 system to another; what works for the shallow, aromatic cradle of CRBN often fails when applied to the deep, hydrophobic groove of VHL. By synthesizing matched warheads with either PEG or alkyl tethers, and projecting the resulting degradation profiles onto ternary-complex cryo-EM envelopes, these studies have derived heuristic rules that are now guiding next-generation efforts—rules that favor solubility for CRBN, but prioritize directional rigidity for VHL to ensure the asparagine-hydroxyl clamp that underlies its substrate recognition.
CRBN-Based PROTACs: PEG vs Alkyl Linkers
The first generation of CRBN-targeted PROTACs used alkyl chains as linkers as they are easy to synthesize and do not stand out metabolically. However, when plotting cellular activity against aqueous solubility it quickly became clear that there was an exponential negative correlation. The alkyl-heavy linkers self-associated to form colloidal aggregates, sequestering the molecules away from the cytosolic compartment, thus reducing the free concentration available for formation of the ternary-complex. Addition of ethylene glycol (PEG) blocks in place of the alkyl chains prevented micellisation by hydrating the linker, but this came at the expense of conformational entropy: a PEG chain can occupy hundreds of extended and collapsed states, which reduces the proportion of geometries that place the key lysine in proximity to the ubiquitination machinery. The key advance was to combine alkynyl linkers (short, rigid alkyl chains next to the CRBN binder to maintain the geometry of the histidine-tryptophan clamp) and a short PEG block next to the warhead to ensure aqueous dispersibility. These compounds were still in the nanomolar range for degradation potency (matching the all-alkyl series), but they formed clear solutions in aqueous buffers and had reduced nonspecific binding to plasticware. In addition, the PEG block could be tuned for desired kidney clearance, without any modification to the aromatic portion, proving that solubility and potency are not mutually exclusive, when the linker is designed as a segmented, rather than a continuous, entity.
The VHL ligase incorporates a shallow, oxygen-dependent pocket that binds hydroxylated proline analogs with the support of three backbone hydrogen bonds and a neighboring asparagine clamp. A linker that imparts torsional stress to this brittle interface can inadvertently misalign the strictly defined hydroxyl geometry and turn a high-affinity recruiter into a poor binder. As such, VHL-targeted degraders are designed with semi-rigid linkers that serve more as a molecular splint rather than an entropic shock absorber. Aromatic or bicyclic units tethered directly to the VHL ligand effectively brace the prolyl surrogate into the bioactive orientation, and a short, freely rotating hinge unit (often just a single ethylene spacer) imparts a modicum of give to absorb small perturbations at the target protein surface. Cryo-EM analyses show that highly flexible PEG spacers permit the ligase to slide away from the catalytic lysine, leading to transient complexes that refresh the unmodified substrate at a rate faster than the E2 can donate ubiquitin. In contrast, completely rigid rods enforce a fixed angle that is not necessarily compatible with every conformational state of the target kinase; as such, iterative structure–activity maturation cycles therefore led to the introduction of "kinkable" units such as para-substituted phenyls or piperidones whose half-chair flexibility serves as a mechanical fuse, allowing them to bend under steric pressure while resisting torsion under non-stressed conditions. The resulting degraders show enhanced residence times and selectivity, underscoring the principle that VHL linkers must serve as directional struts rather than passive tethers if the fragile asparagine clamp is to be maintained throughout the lifetime of the ternary complex.
Best Practices and Design Guidelines
On campaigns where PROTACs have worked, linker design is no longer an after-thought. Instead, the "reach space" between the E3 catalytic face and the lysine-rich zone of the target is mapped out first. Spacer chemistries that satisfy that distance requirement are then selected that also are compatible with membrane transit and metabolic stability. The emerging rule set is therefore tri-partite: match the linker class to the architectural personality of the ligase (shallow CRBN prefers solvated flexors, deep VHL demands directional struts), enforce a conformational bias that places the neo-substrate in ubiquitination range without over-freezing global flexibility, and build in early ADME sentinels—polarity caps, soft ester lability, shape-embedded efflux avoidance—so that potency and exposure are improved in parallel rather than traded off in sequence.
Matching Linker Type to E3 Ligase and Target Distance
CRBN features a solvent-exposed β-hairpin, which His-Trp clamp accommodates a wide range of incoming geometries; in this case, PEG or short alkyl spacers serve as entropic shock absorbers, letting the target rotate until a productive pose is achieved. VHL, by contrast, nestles its hydroxylated ligand in a narrow cleft flanked by Tyr98 and a rigid asparagine ladder; any linker that introduces torsion to this interface is liable to perturb the delicate H-bond network. Semi-rigid aromatics or spiro-cyclized segments are therefore situated adjacent to the VHL ligand, and a single rotatable hinge further along the chain accommodates breathing motions of the POI. Distance estimation can start from cryo-EM or Rosetta-generated ensembles that measure Cα-to-Cα separations across the predicted trimer; the linker is then designed to sample its most probable end-to-end length within ± one bond of that span. Under-short tethers produce steric compression that accelerates dissociation, while over-lengthy ones permit the catalytic lysine to drift out of E2 reach. A practical compromise is a chimeric architecture—rigid segment for orientation, flexible segment for reach—whose length is scanned in single-atom increments until a plateau in degradation efficacy is observed. This plateau, rather than the absolute IC50, is taken as the geometric optimum because it reflects the statistical majority of productive encounters rather than the tightest binary interaction.
Balancing Efficiency, Selectivity, and Pharmacokinetics
These gains are easily offset by off-target effects if the molecule is actively pumped out of cells or passively sequestered in compartments where the UPS is low. Early introduction of soft metabolic handles (ester or carbonate motifs that collapse to polar acids after CYP oxidation) limits intracellular buildup of lipophilic fragments that might compete for ternary complex. Selectivity is dictated by incorporating shape-cliffs into the linker itself: an ortho-methyl on an aromatic ring can twist the π-stack to the point where homologous kinases that lack the complementary leucine shelf are rejected, giving isoform selectivity without altering the high-affinity warhead. Polar surface area is dialed in with rigidity; rigid spacers can support higher polarity because they pay the desolvation penalty once, while flexible chains need to be kept below a running tally to avoid entropic rain-out. Permeability is assessed in parallel with artificial membrane assays and then verified with intact-cell uptake studies that quantify unbound fraction by equilibrium dialysis; any compound with a cytosolic free level below ternary-complex Kd is reformulated before being advanced to animal studies. The iterative loop is closed by re-introducing top performers into the modelling ensemble, re-weighting the conformer landscape and kicking off another round of micro-scale synthesis until the degradation signature, off-target profile and exposure metrics are all converged in the same, drug-like window.
Our Linker and PROTAC Intermediate Solutions
Ready-to-Use Linker Libraries and Building Blocks
We offer a curated range of ready-to-use PROTAC linker libraries and chemical building blocks designed to streamline your discovery workflow. Our linkers are available in varied lengths, polarities, and conformations - including alkyl, PEG, and aromatic spacers - to support structure–activity optimization and ternary complex stability. Each compound is manufactured under stringent quality standards and supplied with complete HPLC, LC-MS, and NMR documentation, ensuring reproducibility across research environments. From rapid screening to late-stage optimization, our linker inventory enables faster and more reliable PROTAC development.
Custom Synthesis Tailored to Your Target Molecule
When standard linkers don't meet your design requirements, our chemistry team provides custom synthesis services for target-specific linker creation. We collaborate closely with your scientists to engineer linkers that achieve the ideal length, rigidity, and functional balance for your E3 ligase-target pair. Our team can introduce specialized features - such as click-chemistry handles, cleavable motifs, or hydrophobic tuning groups - while maintaining excellent solubility and stability profiles. Every project includes feasibility assessment, synthetic route design, and analytical validation to ensure a smooth transition from concept to delivery. With our tailor-made linker synthesis, you can precisely fine-tune PROTAC efficiency and selectivity for your specific biological system.
Technical Support for Design, Scale-Up, and Purification
Beyond synthesis, we provide end-to-end technical support covering every stage of your PROTAC linker project. Our scientists assist with:
- Design consultation to optimize linker geometry and physicochemical balance.
- Scale-up strategies to move from milligram research batches to pilot-scale production.
- Purification optimization, including HPLC method development and impurity profiling.
This integrated service ensures your linkers and intermediates are ready for immediate biological evaluation or further conjugation. By combining synthetic capability with analytical insight, we help transform promising PROTAC concepts into validated chemical tools.
Accelerate Your PROTAC Development with Expert Linker Solutions
Browse our online catalog of validated PROTAC linkers and intermediates, organized by length, polarity, and functional type. Each entry includes analytical specifications, availability, and datasheets, enabling quick compound selection for your next project. Explore our catalog now to find linkers that fit your PROTAC design strategy and degradation goals.
Need something unique? Reach out to our R&D chemistry team for custom linker synthesis tailored to your target, E3 ligase, and desired molecular properties. We provide rapid quotations, technical feedback, and delivery timelines, ensuring smooth coordination from inquiry to fulfillment. Contact us today to discuss your project and gain access to expert-designed PROTAC linker solutions built for scientific and commercial success.
FAQs
1. Why is linker optimization essential in PROTAC design?
The linker controls spatial positioning between the target and E3 ligase, influencing binding stability and degradation selectivity.
2. What types of linkers are commonly used?
Alkyl, PEG, and aromatic linkers are typical; each offers different flexibility and hydrophobicity profiles.
3. How do researchers select the ideal linker?
They evaluate degradation potency, cell permeability, and computational modeling to find the best structure-activity balance.
References
- Image retrieved from Figure 1 " The PROTACs mode of action, involving ubiquitination and eventual degradation of POI.," Anwar Z.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Anwar Z, Ali M S, Galvano A, et al. PROTACs: the future of leukemia therapeutics[J]. Frontiers in Cell and Developmental Biology, 2022, 10: 851087. https://doi.org/10.3389/fcell.2022.851087.
- Yedla P, Babalghith A O, Andra V V, et al. PROTACs in the management of prostate cancer[J]. Molecules, 2023, 28(9): 3698. https://doi.org/10.3390/molecules28093698.
- Luo Y, Song D, Zhang C, et al. ProLinker–Generator: Design of a PROTAC Linker Base on a Generation Model Using Transfer and Reinforcement Learning[J]. Applied Sciences, 2025, 15(10): 5616. https://doi.org/10.3390/app15105616.
