Approved PROTAC Drugs: Mechanism, Clinical Pipeline, Assay Development, and Future Directions
* Please be kindly noted that our services and products can only be used for research to organizations or companies and not intended for any clinical or individuals.
Consult with Our Experts
Article
What are PROTACs?
Proteolysis-targeting chimeras, or PROTACs, are heterobifunctional molecules designed to remove disease-driving proteins from cells. Unlike conventional inhibitors, which usually block a protein’s active site or signaling function, PROTACs induce degradation of the entire target protein. A typical PROTAC contains three parts: a ligand that binds the protein of interest, a linker, and a ligand that recruits an E3 ubiquitin ligase. When these elements work together, the PROTAC brings the target protein and E3 ligase into proximity, enabling ubiquitination and proteasomal degradation of the target.
This mechanism gives PROTACs several potential advantages. Because the target protein is removed rather than simply inhibited, PROTACs may suppress enzymatic, scaffolding, transcriptional, and protein-protein interaction functions at the same time. They may also be useful for targets that are difficult to inhibit with traditional small molecules, including transcription factors, mutant signaling proteins, and proteins with no obvious catalytic pocket. However, PROTACs also bring drug-development challenges: they are often larger than classical small molecules, may fall outside conventional “rule-of-five” property space, and require optimization of ternary complex formation, intracellular exposure, degradation kinetics, and pharmacodynamic durability.
Vepdegestrant (Veppanu): The First FDA-Approved PROTAC Drug
Vepdegestrant, formerly known as ARV-471 and marketed as VEPPANU, is the first FDA-approved PROTAC drug. It was approved in 2026 for adults with estrogen receptor-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer whose disease progressed after at least one line of endocrine therapy. The approval is important not only for breast cancer treatment, but also for the broader targeted protein degradation field, because it provides the first regulatory validation that a PROTAC can become an approved medicine.
Table 1. Vepdegestrant at a Glance — Key Parameters of the First Approved PROTAC.
| Item | Key information |
|---|
| Drug name | Vepdegestrant / VEPPANU |
| Modality | Oral heterobifunctional PROTAC degrader |
| Target | Estrogen receptor |
| E3 ligase | Cereblon |
| Approved use | ER+/HER2−, ESR1-mutated advanced or metastatic breast cancer after endocrine therapy |
| Pivotal trial | VERITAC-2 |
| Key efficacy result | Median PFS: 5.0 months vs 2.1 months with fulvestrant |
The approved product is described in its prescribing information as a heterobifunctional protein degrader composed of an estrogen receptor-binding domain joined by a linker to an E3 ligase-binding domain. It binds estrogen receptor and cereblon, causing cereblon-mediated polyubiquitination and proteasomal degradation of estrogen receptor. The molecule has a molecular weight of 723.90 Daltons, illustrating that clinically successful oral PROTACs can extend beyond the physicochemical space of many traditional oral small molecules.
Vepdegestrant for ER+/HER2- ESR1- Mutated Advanced Breast Cancer
The approved population is biomarker-defined: patients must have ER-positive, HER2-negative advanced or metastatic breast cancer with an ESR1 mutation detected by an FDA-authorized test. ESR1 mutations are clinically relevant because they can support estrogen receptor activity even when estrogen signaling is therapeutically suppressed. In this setting, degrading the receptor protein itself may be more direct than only blocking receptor activation.
Vepdegestrant is designed to degrade both wild-type and mutant estrogen receptor. The label states that it induced degradation of wild-type and mutant ER, inhibited ER-dependent breast cancer cell-line proliferation in vitro, and demonstrated antitumor activity in breast cancer models containing wild-type or mutant ESR1.
VERITAC-2 Phase 3 Trial Results and Clinical Outcomes
VERITAC-2 was a randomized, open-label, active-controlled, multicenter Phase 3 trial enrolling 624 adults with ER-positive, HER2-negative advanced or metastatic breast cancer; 270 of these patients had ESR1-mutated tumors. Patients had disease progression after one to two prior lines of endocrine therapy, including prior CDK4/6 inhibitor exposure.
In the ESR1-mutated population, vepdegestrant improved progression-free survival compared with fulvestrant. Median PFS was 5.0 months with vepdegestrant versus 2.1 months with fulvestrant, with a hazard ratio of 0.57. The confirmed objective response rate was 19% versus 4% among patients with measurable disease. Overall survival was immature at the time of the PFS analysis.
PROTAC Drugs in Clinical Trials: The Expanding Pipeline
The clinical PROTAC pipeline is expanding across oncology, immunology, inflammation, and neurology. Oncology remains the most advanced area, especially hormone receptor-driven tumors, B-cell malignancies, and genetically defined solid tumors. Non-oncology programs are also advancing, particularly where degradation may silence transcriptional or immune signaling pathways that are difficult to inhibit with classical small molecules.
Table 2. PROTAC Degraders in Clinical Development — Selected Pipeline Overview.
| Candidate | Target | Lead indication area | Stage | E3 ligase | Inquiry |
|---|
| Vepdegestrant | ER | Breast cancer | Approved | CRBN | Inquiry |
| CC-94676 | AR | Prostate cancer | Phase 3 | CRBN | Inquiry |
| ASP3082 / setidegrasib | KRAS
G12D | Pancreatic cancer / solid tumors | Phase 3 | VHL | Inquiry |
| BGB-16673 | BTK | CLL/SLL | Phase 3 | Not fully disclosed | Inquiry |
| NX-5948 | BTK | CLL, NHL, WM | Phase 1/2 | CRBN | Inquiry |
| ARV-766 | AR | Prostate cancer | Phase 2 | CRBN-based | Inquiry |
| KT-621 | STAT6 | Atopic dermatitis, asthma | Phase 2b | Not disclosed | Inquiry |
| KT-333 | STAT3 | Hematologic malignancies | Phase 1 | VHL | Inquiry |
| PRT3789 | SMARCA2 | SMARCA4-mutant tumors | Phase 1 | VHL | Inquiry |
This table is intentionally selective rather than exhaustive. It focuses on representative approved, late-stage, and mechanistically important clinical programs. Some E3 recruiters remain undisclosed in public materials, and clinical status can change quickly.
PROTAC Degraders in Phase 3 Clinical Development
Three areas now define late-stage PROTAC development: estrogen receptor degradation, androgen receptor degradation, and degradation of resistance-associated or mutation-defined oncology targets. CC-94676 is being evaluated in a Phase 3 study for metastatic castration-resistant prostate cancer after prior androgen receptor pathway therapy. It is described as a dual androgen receptor ligand-directed degrader and antagonist, which means it is designed to both degrade AR and antagonize AR signaling.
ASP3082, also known as setidegrasib, is a KRAS G12D-targeted protein degrader. A Phase 3 study is evaluating it in combination with chemotherapy regimens for first-line KRAS G12D-mutated metastatic pancreatic ductal adenocarcinoma, with overall survival as a primary endpoint. Public structural work has shown that ASP3082 forms a KRAS G12D-degrader-VHL ternary complex and was optimized through structure-guided design.
BGB-16673 is a BTK degrader being evaluated in Phase 3 for relapsed or refractory CLL/SLL after prior exposure to BTK and BCL2 inhibitors. This reflects a broader trend in hematologic malignancies: degrading BTK may address both kinase-dependent signaling and non-catalytic functions that can remain relevant after inhibitor resistance.
PROTAC Degraders in Phase 2 Clinical Development
Phase 2 programs are broadening the clinical scope of PROTACs. ARV-766 is a next-generation androgen receptor degrader in prostate cancer, designed to improve on earlier AR degrader concepts and cover clinically relevant resistance biology. BTK degradation is also moving beyond early dose escalation, with NX-5948 being studied in CLL and other B-cell malignancies. Bexobrutideg, also known as NX-5948, is described as an orally administered small-molecule BTK degrader that removes wild-type and mutant BTK through cereblon-mediated ubiquitination and proteasomal degradation.
Inflammatory disease is another important Phase 2 frontier. KT-621 is an oral STAT6 degrader being studied in atopic dermatitis and asthma. STAT6 is a transcription factor downstream of IL-4 and IL-13 signaling, making it a central node in type 2 inflammation. Phase 2b studies in atopic dermatitis and asthma are intended to test whether oral degradation of STAT6 can compete with biologic pathway blockade in chronic inflammatory disease.
PROTAC Degraders in Phase 1 Clinical Development
Phase 1 programs show how broad the modality could become. KT-333 is a STAT3 degrader recruiting VHL, designed for malignancies where STAT3 signaling contributes to survival, immune evasion, or oncogenic transcription. PRT3789 targets SMARCA2 in SMARCA4-mutant tumors, using synthetic lethality as its therapeutic rationale. In both cases, degradation is intended to reach proteins where conventional inhibition may be difficult or insufficient.
Early clinical studies also include degraders against BCL6, BCL-XL, mutant kinases, epigenetic regulators, and neurological disease targets. These programs are important because they test whether PROTAC pharmacology can be applied beyond the first validated hormone-receptor and kinase examples. The central question is no longer whether PROTACs can work at all, but where degradation provides a meaningful advantage over inhibition.
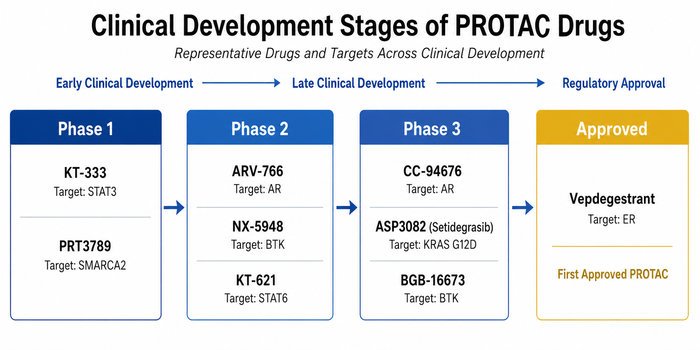 Fig.1 PROTAC drug pipeline from Phase 1 candidates to the first approved PROTAC therapy (BOC Sciences Original).
Fig.1 PROTAC drug pipeline from Phase 1 candidates to the first approved PROTAC therapy (BOC Sciences Original).
Ready to Advance Your PROTAC Drug Toward the Clinic?
BOC Sciences supports PROTAC discovery and development with integrated solutions covering degrader design, synthesis, target degradation assays, and pharmacokinetic evaluation.
Get Consultation
Structural Features and Structure-Activity Relationships of Clinical PROTAC Drugs
PROTAC structure-activity relationships are more complex than classical inhibitor SAR. A degrader must bind two proteins, form a productive ternary complex, induce ubiquitination, enter the right cells and tissues, and maintain adequate exposure. Small changes to the linker, E3 ligand, or target ligand can dramatically alter potency, selectivity, pharmacokinetics, and degradation depth.
Table 3. Key Structural Features of Approved and Clinical-Stage PROTACs.
| Candidate | Route | Key structural feature |
|---|
| Vepdegestrant | Oral | ER ligand-linker-CRBN ligand architecture |
| CC-94676 | Oral | AR degradation plus antagonism |
| ASP3082 | IV | Structure-guided KRAS G12D ternary complex |
| NX-5948 | Oral | BTK degrader with CNS penetration potential |
| KT-333 | Systemic | Transcription-factor degrader |
| PRT3789 | IV | Synthetic-lethal SMARCA2 degrader |
The most important structural lesson from clinical PROTACs is that whole-molecule behavior matters. Binary affinity alone is not enough. A target ligand can be potent but fail to support productive ternary geometry. An E3 ligand can recruit a ligase but also create safety or selectivity liabilities. A linker can improve degradation at one length and destroy activity at another. Ternary-complex stability and cooperativity often correlate with cellular degradation potency and degradation rate.
E3 Ligase Ligand Features in Clinical PROTACs
Most clinical PROTACs still rely on a small number of E3 ligase systems, especially CRBN and VHL. CRBN-based degraders have shown oral drug potential in ER, AR, and BTK programs. VHL-based degraders are prominent in structural biology-driven programs, including KRAS G12D, STAT3, SMARCA2, and BCL-XL degraders. E3 ligase choice influences potency, tissue activity, degradation selectivity, and safety. It also affects the ternary complex interface. For example, ASP3082 optimization used the KRAS G12D-degrader-VHL ternary complex to improve degradation activity and selectivity, showing how E3-ligand design can shape the final protein-protein interface rather than merely recruit a ligase.
PROTAC Linker Features and Optimization
The linker is not just a spacer. It determines distance, orientation, conformational flexibility, polarity, and the probability of productive ternary-complex formation. A linker that is too short may prevent the two proteins from forming a stable interface. A linker that is too long or too flexible may increase entropic cost, reduce cooperativity, worsen permeability, and increase metabolic liability. Clinical optimization often tests multiple linker lengths, attachment points, and chemical motifs. The goal is to produce a molecule that is potent in cells, selective across the proteome, stable enough for in vivo exposure, and compatible with the intended route of administration. In the ASP3082 program, linker modification based on ternary-complex modeling contributed to improved KRAS G12D degradation potency, demonstrating how linker SAR can directly improve target degradation.
Target Protein Ligand Features in Clinical PROTACs
The target-binding ligand in a PROTAC does not always need to be a perfect inhibitor. It needs to bind the target in a way that supports productive E3 ligase recruitment and ubiquitination. This distinction is important for targets where complete inhibition is difficult but target engagement is possible. Hormone receptor degraders use receptor-binding ligands to recruit ER or AR into proximity with an E3 ligase. BTK degraders use kinase-binding motifs but aim to remove BTK protein rather than simply block catalytic activity. KRAS G12D degraders use mutant-selective binding elements to recruit a disease-driving mutant protein into a degradative ternary complex. In each case, the best target ligand is the one that supports degradation, not necessarily the one that looks strongest in a traditional inhibitor screen.
Table 4. Services for PROTAC Design and Structural Optimization.
| Service Name | Description | Inquiry |
|---|
| PROTAC Library | Provides ready-to-screen degrader molecules for early target validation, hit discovery, and preliminary structure-activity relationship studies. | Inquiry |
| Linker Library | Supports rapid exploration of linker length, polarity, rigidity, flexibility, and attachment geometry to optimize ternary complex formation and degradation activity. | Inquiry |
| Warhead Library | Provides target-binding building blocks for designing PROTAC protein-of-interest ligands and accelerating degrader construction. | Inquiry |
| Design of the Ligase System | Helps select and design suitable E3 ligase recruitment strategies based on target biology, tissue context, degradation mechanism, and developability requirements. | Inquiry |
| PROTAC Design Services | Provides integrated PROTAC design support, including target ligand selection, E3 ligase recruiter selection, linker strategy, and overall degrader architecture optimization. | Inquiry |
| Linker Design and Optimization Services | Optimizes linker length, composition, rigidity, polarity, and exit vectors to improve degradation potency, selectivity, permeability, and pharmacokinetic properties. | Inquiry |
| Linker Binding Site Selection and Design | Identifies suitable attachment sites on target ligands and E3 ligase ligands to support productive ternary complex formation and efficient ubiquitination. | Inquiry |
| Ligand Design for Target Protein | Supports the discovery or optimization of protein-of-interest ligands that can serve as effective warheads for PROTAC degrader development. | Inquiry |
PROTAC Assay Development and Evaluation Methods
Unlike conventional small-molecule inhibitors, PROTAC drugs do not rely only on target binding or enzymatic inhibition. Their activity depends on whether they can recruit an E3 ubiquitin ligase, form a productive ternary complex, induce target protein ubiquitination, and ultimately trigger proteasome-mediated degradation. Therefore, PROTAC evaluation should cover the full workflow from molecular binding and ternary complex formation to cellular degradation, in vivo pharmacokinetics, and pharmacodynamic validation.
PROTAC Ternary Complex Formation Assays
Because a PROTAC is a bifunctional molecule composed of a target protein ligand, a linker, and an E3 ligase ligand, its core mechanism depends on the formation of a ternary complex among the target protein, the PROTAC, and the E3 ubiquitin ligase. Therefore, early evaluation should not only determine whether the PROTAC can bind the target protein or the E3 ligase separately, but also whether it can bridge both proteins at the same time and form a stable complex capable of supporting degradation.
Ternary complex formation is commonly evaluated using general biophysical or biochemical methods. Techniques such as surface plasmon resonance, isothermal titration calorimetry, microscale thermophoresis, fluorescence polarization, fluorescence resonance energy transfer-based assays, and other proximity-based assays can be used to analyze the binding behavior and complex stability among the PROTAC, target protein, and E3 ligase. These assays commonly assess binding affinity, complex formation efficiency, dissociation rate, and cooperativity.
Cooperativity is an important parameter in PROTAC evaluation. If a PROTAC induces favorable protein-protein interactions between the target protein and the E3 ligase, ternary complex stability may increase, thereby improving degradation efficiency. Conversely, if the ternary complex geometry is unfavorable, even a PROTAC with strong binary binding to both proteins may fail to produce effective degradation. Therefore, ternary complex formation assays provide important guidance for linker design, E3 ligase selection, and overall molecular optimization.
PROTAC Hook Effect Assessment
A PROTAC must bind both the target protein and the E3 ligase to form an effective ternary complex, so its concentration-response relationship may differ from that of a conventional inhibitor. At an appropriate concentration, the PROTAC bridges the target protein and E3 ligase, promoting ubiquitination and degradation. However, at excessively high concentrations, the PROTAC may separately saturate the target protein and the E3 ligase, forming too many binary complexes and reducing productive ternary complex formation. This phenomenon is commonly known as the hook effect.
For this reason, PROTAC activity evaluation should assess whether the candidate molecule shows a bell-shaped concentration-response curve. In experimental design, a broad concentration gradient is usually recommended rather than a single concentration or a narrow dose range. By comparing ternary complex formation, ubiquitination levels, and target protein degradation at different concentrations, researchers can determine whether activity decreases at high compound concentrations.
Hook effect assessment is important for dose selection and interpretation of pharmacological activity. If a candidate PROTAC shows strong degradation activity at low or moderate concentrations but reduced activity at high concentrations, its effective concentration window should be further defined. For in vivo studies, pharmacokinetic data should also be used to determine whether actual exposure falls within the effective degradation range, helping avoid misinterpretation caused by underdosing or overdosing.
Target Protein Degradation Assays
Because the final pharmacological goal of a PROTAC is to reduce the level of the target protein rather than simply block its activity, target protein degradation assays are central to PROTAC evaluation. Even if a candidate molecule can bind both the target protein and the E3 ligase, it must still be shown to induce actual target protein degradation inside cells.
Target protein degradation is usually assessed in cellular systems. Common methods include Western blotting, ELISA, cellular immunofluorescence, flow cytometry, quantitative proteomics, and reporter-based cellular assay systems. These methods can be used to measure changes in target protein levels across different PROTAC concentrations and treatment times. Common evaluation parameters include DC50, Dmax, degradation rate, time to maximum degradation, and target protein recovery after compound washout.
Mechanistic validation experiments are also needed to confirm that target protein reduction is mediated by the intended PROTAC mechanism. For example, proteasome inhibition, E3 ligase competition, target ligand competition, or E3 ligase knockdown/knockout models can be used to determine whether degradation depends on the ubiquitin-proteasome system and the selected E3 ligase. Ubiquitination assays can further confirm whether the target protein undergoes ubiquitin modification after PROTAC treatment.
Selectivity evaluation is also important. Because PROTACs induce protein degradation through E3 ligase recruitment, they may carry a risk of degrading unintended proteins. Therefore, during candidate optimization, proteomic analysis is often used to evaluate the impact of the PROTAC on proteins other than the intended target, supporting safety and selectivity assessment.
In Vivo Pharmacokinetic Evaluation of PROTAC Degraders
PROTAC molecules often have relatively high molecular weights and complex structures, and their physicochemical properties may differ from those of traditional oral small-molecule drugs. Therefore, even if a PROTAC shows strong degradation activity in vitro, its absorption, distribution, metabolism, and excretion properties must be evaluated in vivo. Pharmacokinetic evaluation helps determine whether the candidate molecule can achieve sufficient exposure and tissue distribution to support sustained target protein degradation.
Pharmacokinetic studies commonly assess the plasma concentration-time profile, maximum plasma concentration, time to maximum concentration, area under the curve, half-life, clearance, bioavailability, tissue distribution, and protein binding. For orally administered PROTACs, solubility, gastrointestinal absorption, metabolic stability, and food effects are particularly important. For PROTACs intended to act in the central nervous system, brain exposure and blood-brain barrier penetration should also be evaluated.
Pharmacokinetic results should be interpreted together with in vitro activity data. A PROTAC with strong in vitro activity may fail to reach effective degradation concentrations in vivo if exposure is insufficient or clearance is rapid. Conversely, a PROTAC with moderate in vitro activity may still produce sustained pharmacological effects if it has a longer half-life or favorable tissue distribution. Therefore, PK evaluation is a key step in determining whether a PROTAC candidate has sufficient potential for further development.
Pharmacodynamic Evaluation of PROTAC Degraders
Because the intended outcome of PROTAC treatment is target protein reduction, pharmacodynamic evaluation should answer two main questions: whether the candidate molecule truly induces target degradation in vivo, and whether that degradation leads to downstream pathway modulation or disease-relevant biological effects. Therefore, PROTAC pharmacodynamic studies usually measure target protein levels, downstream signaling changes, and functional outcomes in disease models.
In oncology research, pharmacodynamic samples may be collected from tumor tissue, peripheral blood cells, circulating tumor cells, or other relevant tissues. Detection methods may include Western blotting, immunohistochemistry, ELISA, flow cytometry, quantitative PCR, transcriptomic analysis, or proteomic analysis. Common evaluation endpoints include the degree of target protein degradation, pathway inhibition, changes in cell proliferation, apoptosis markers, and tumor growth inhibition.
Pharmacodynamic evaluation should also consider the time dimension. Because PROTACs induce protein degradation, the duration of pharmacological effect depends not only on drug concentration but also on target protein resynthesis, tissue exposure duration, and pathway recovery. Therefore, in vivo studies commonly include multiple sampling time points to monitor target degradation and recovery, providing support for dosing frequency and dose-regimen design.
Table 5. Recommended Services for PROTAC Evaluation at BOC Sciences.
| Service Name | Description | Inquiry |
|---|
| PROTAC In Vitro Evaluation | Evaluates PROTAC performance in biochemical and cellular systems, including target engagement, degradation potency, selectivity, and mechanism validation. | Inquiry |
| PROTAC In Vitro Metabolism | Assesses metabolic stability, metabolite formation, and degradation pathways in liver microsomes, hepatocytes, or other relevant in vitro systems. | Inquiry |
| PROTAC High-Throughput Screening | Screens PROTAC libraries or candidate degraders to rapidly identify active compounds with target degradation or pathway-modulating activity. | Inquiry |
| Degradation Ability Assay | Measures target protein degradation efficiency, including DC50, Dmax, degradation kinetics, and recovery after compound washout. | Inquiry |
| PROTAC Activity Assay | Evaluates functional activity of PROTAC candidates by measuring target degradation, downstream pathway inhibition, or disease-relevant cellular responses. | Inquiry |
| Solubility and Stability | Determines aqueous solubility, chemical stability, plasma stability, and storage stability to support developability assessment. | Inquiry |
| E3 Ubiquitin Ligase Activity Assay | Assesses E3 ligase recruitment and ubiquitination activity to confirm that the selected ligase system can support target protein degradation. | Inquiry |
| Binding Affinity Measurement | Measures binding affinity between the PROTAC and target protein, E3 ligase, or ternary complex components using biochemical or biophysical methods. | Inquiry |
| PROTAC Ternary Complex Assay | Evaluates formation, stability, and cooperativity of the target-PROTAC-E3 ligase ternary complex, a key determinant of degradation efficiency. | Inquiry |
| PROTAC In Vivo Evaluation | Assesses pharmacokinetics, pharmacodynamics, target degradation, efficacy, and safety-related endpoints in relevant animal studies. | Inquiry |
| PROTAC In Vivo Animal Model | Establishes or uses disease-relevant animal models to evaluate PROTAC exposure, tissue distribution, target degradation, and therapeutic response. | Inquiry |
| Protein-ligand Structure Analysis | Analyzes protein-ligand binding modes and structural interactions to guide warhead design, linker placement, and PROTAC optimization. | Inquiry |
Overall, PROTAC pharmacodynamic evaluation should be integrated with pharmacokinetic data to establish a PK/PD relationship. By analyzing the relationship among in vivo exposure, target protein degradation, and therapeutic response in disease models, researchers can more accurately determine the effective dose range, duration of action, and development value of a candidate PROTAC.
PROTAC Targets and Therapeutic Application Areas
PROTACs are being applied to targets where removing the protein may be more valuable than inhibiting one function. The most advanced areas are hormone receptors, kinases, mutant oncogenic signaling proteins, transcription factors, and proteins involved in immune signaling.
PROTACs Targeting Hormone Receptors in Breast and Prostate Cancer
Hormone receptors are among the best validated PROTAC targets. In breast cancer, vepdegestrant shows that degrading estrogen receptor can provide clinical benefit in ESR1-mutated disease after endocrine therapy. In prostate cancer, AR degraders are being developed to address tumors that remain dependent on androgen receptor biology after androgen receptor pathway inhibition. The scientific rationale is straightforward: if receptor signaling remains central to disease progression, removing the receptor protein may suppress both ligand-dependent and ligand-independent activity. This is especially relevant when mutations, overexpression, or altered receptor function reduce the effectiveness of conventional antagonists.
PROTACs Targeting BTK and Kinases in Hematologic Malignancies
BTK degraders are designed to remove BTK protein rather than simply inhibit kinase activity. This distinction may matter in relapsed or refractory B-cell malignancies, where resistance to covalent and non-covalent BTK inhibitors can emerge. A degrader may suppress catalytic signaling and non-catalytic scaffolding functions at the same time. NX-5948 and BGB-16673 illustrate this strategy. NX-5948 is described as degrading wild-type and mutant BTK through cereblon-mediated ubiquitination, while BGB-16673 is being evaluated in Phase 3 CLL/SLL studies after prior BTK and BCL2 inhibitor exposure.
PROTACs for Previously Undruggable Targets: KRAS, STAT3, and p53
KRAS G12D degradation is one of the most closely watched examples of PROTAC expansion into historically difficult targets. ASP3082 selectively degrades KRAS G12D through a VHL-recruited ternary complex and has moved into late-stage clinical evaluation for KRAS G12D-mutated pancreatic cancer. STAT3 is another challenging target because it acts as a transcription factor and signaling hub rather than a conventional enzyme. STAT3 degrader programs test whether targeted degradation can succeed where direct inhibition has been difficult. p53-related degrader strategies remain more experimental, especially because p53 biology is context-dependent: some tumors require stabilization of wild-type p53, while others may benefit from degradation or functional suppression of mutant p53.
PROTACs Beyond Oncology: Neurodegenerative and Autoimmune Diseases
Beyond oncology, PROTACs are being explored in autoimmune, inflammatory, neurological, and neuromuscular disorders. STAT6 degradation is a leading example in type 2 inflammatory disease, where an oral degrader could offer a different therapeutic profile from injectable biologics or broad immunosuppressive agents. Neurological applications require additional design features, especially brain penetration, selectivity, and long-term safety. For chronic diseases, the benefit-risk threshold is higher than in late-line oncology. A successful non-oncology PROTAC must combine strong target validation, tissue-appropriate exposure, durable pharmacodynamics, and a safety profile suitable for repeated or long-term dosing.
Table 6. BOC Sciences PROTAC Solutions for Diverse Therapeutic Applications.
| Service Name | Description | Inquiry |
|---|
| PROTACs for Autoimmune Diseases | Supports the development of PROTAC degraders targeting immune-related signaling proteins and inflammatory pathways for autoimmune disease research. | Inquiry |
| PROTACs for Lymphoma | Provides PROTAC design and evaluation solutions for lymphoma-associated targets, including kinases, transcription factors, and epigenetic regulators. | Inquiry |
| PROTACs for Prostate Cancer | Supports the discovery and optimization of PROTAC degraders for prostate cancer targets such as androgen receptor and related resistance mechanisms. | Inquiry |
| PROTACs for Leukemia | Offers PROTAC solutions for leukemia research by targeting disease-driving proteins involved in survival, proliferation, and abnormal hematopoietic signaling. | Inquiry |
| PROTACs for Breast Cancer | Supports PROTAC development for breast cancer targets, including hormone receptors and resistance-associated proteins. | Inquiry |
| PROTACs for Neurodegenerative Diseases | Provides PROTAC research solutions for degrading disease-associated proteins involved in neurodegeneration and protein aggregation. | Inquiry |
| PROTACs for Non-small Cell Lung Cancer | Supports PROTAC degrader discovery for NSCLC-related targets, including oncogenic kinases, mutant signaling proteins, and resistance-associated drivers. | Inquiry |
Next-Generation PROTAC Technology and Future Directions
The first approved PROTAC validates the modality, but future success will depend on better design tools, improved tissue selectivity, broader E3 ligase options, and stronger clinical biomarker strategies. The field is moving from proof of concept to product differentiation.
Advanced PROTAC Design: Covalent, Macrocyclic, and Tissue-Targeted Degraders
Next-generation PROTACs may use covalent binding, macrocyclic structures, conformational restriction, or tissue-targeted delivery. Covalent degraders can improve target engagement but must be designed carefully to preserve turnover and avoid excessive off-target reactivity. Macrocyclic and conformationally restricted degraders may improve permeability, ternary-complex geometry, and selectivity.
Tissue-targeted degraders are another promising direction. The goal is to concentrate degradation activity in diseased cells while sparing normal tissue. This may be achieved through targeted delivery, tissue-enriched E3 ligase recruitment, or disease-state-specific biology.
Emerging E3 Ligases and Expanding the PROTAC Toolbox
Although hundreds of E3 ligases exist in human biology, clinical PROTACs still rely heavily on CRBN and VHL. Expanding the usable E3 ligase toolbox could enable more tissue-specific degradation, reduce resistance risks, and improve therapeutic index. Emerging E3 ligases may be selected for tissue expression, subcellular localization, disease-state regulation, or reduced neosubstrate liability.
The main challenge is ligand discovery. Each new E3 ligase requires a high-quality recruiter, structural validation, cellular degradation evidence, and safety assessment. The most useful future E3 ligases will not simply be those with potent recruiters; they will be those that improve clinical selectivity.
The PROTAC Drug Market and Pipeline Outlook
The approval of vepdegestrant marks the transition of PROTACs from an experimental platform to an approved therapeutic class. The next wave of data will determine how broadly the modality can expand. The strongest opportunities are likely to be targets where degradation clearly improves on inhibition: mutant oncogenes, resistance-associated kinases, hormone receptors, transcription factors, and proteins with scaffolding functions.
Future clinical success will require more than potent degradation in cells. Successful PROTACs will need clear patient selection, reliable pharmacodynamic biomarkers, manageable safety, scalable manufacturing, and differentiation from existing therapies. The field now has its first approved medicine; the next challenge is proving that targeted protein degradation can become a repeatable drug-development strategy across multiple diseases.
References
- U.S. Food and Drug Administration. Oncology (Cancer)/Hematologic Malignancies Approval Notifications. Accessed May 8, 2026. https://www.fda.gov/drugs/resources-information-approved-drugs/oncology-cancerhematologic-malignancies-approval-notifications
- ClinicalTrials.gov. A Study of BGB-16673 Compared to Investigator's Choice in Participants With Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma Previously Exposed to Both Bruton Tyrosine Kinase (BTK) and B-cell Leukemia/Lymphoma 2 Protein (BCL2) Inhibitors (CaDAnCe-302). ClinicalTrials.gov identifier: NCT06846671. Accessed May 8, 2026. https://clinicaltrials.gov/study/NCT06846671
- ClinicalTrials.gov. A Study of NX-5948 in Adults With Relapsed/Refractory B-cell Malignancies. ClinicalTrials.gov identifier: NCT05131022. Accessed May 8, 2026. https://clinicaltrials.gov/study/NCT05131022
- ClinicalTrials.gov. A Study of KT-621 Administered Orally to Participants With Moderate to Severe Atopic Dermatitis (BROADEN2). ClinicalTrials.gov identifier: NCT07217015. Accessed May 8, 2026. https://clinicaltrials.gov/study/NCT07217015
- ClinicalTrials.gov. Safety, PK, PD, Clinical Activity of KT-333 in Adult Patients With Refractory Lymphoma, Large Granular Lymphocytic Leukemia, Solid Tumors. ClinicalTrials.gov identifier: NCT05225584. Accessed May 8, 2026. https://clinicaltrials.gov/study/NCT05225584
- ClinicalTrials.gov. PRT3789 Monotherapy and in Combo w/ Docetaxel in Participants w/ Advanced or Metastatic Solid Tumors w/ SMARCA4 Mutation. ClinicalTrials.gov identifier: NCT05639751. Accessed May 8, 2026. https://clinicaltrials.gov/study/NCT05639751
Our Support
PROTAC Drug Development Support at BOC Sciences
BOC Sciences provides integrated PROTAC products and development services, supporting researchers in degrader design, custom synthesis, biological evaluation, and preclinical optimization to accelerate targeted protein degradation research.

PROTAC Products and Compound Libraries
PROTAC Design and Molecular Optimization
PROTAC Synthesis and Analytical Characterization
- Custom synthesis of PROTAC molecules, intermediates, warheads, linkers, and E3 ligase ligand-linker conjugates
- Synthesis of PROTAC analog series to support structure-activity relationship and structure-degradation relationship studies
- Chemical characterization and quality control using suitable analytical methods to confirm compound identity, purity, and consistency
PROTAC Evaluation and Preclinical Support
- In vitro evaluation of target degradation, ternary complex formation, ubiquitination, binding affinity, activity, solubility, stability, and metabolism
- In vivo evaluation of pharmacokinetics, pharmacodynamics, tissue distribution, target degradation, and therapeutic response
- Disease-relevant animal model studies and integrated PK/PD analysis to support preclinical PROTAC development and candidate advancement
Frequently Asked Questions (FAQ)

Still have questions?
Contact Us
Are There Any Approved PROTAC Drugs Today?
Yes. Current public reports identify vepdegestrant/VEPPANU as the first approved PROTAC drug, marking a major validation point for targeted protein degradation as a drug discovery modality. For developers, this milestone shifts PROTACs from a high-potential research concept toward a more commercially visible platform. It also increases interest in degrader design, target selection, E3 ligase recruitment, linker strategy, and developability optimization across new discovery programs.
What Targets Are Suitable for PROTAC Degradation?
Suitable PROTAC targets usually have disease relevance, a ligandable binding site, measurable cellular expression, and the potential to be efficiently recruited to an E3 ligase for degradation. Customers often ask this question because target selection strongly influences the difficulty, cost, and success probability of a degrader program. BOC Sciences can support early PROTAC projects through target protein services, ligand evaluation, E3 ligase strategy, linker design, and degrader optimization, helping developers assess whether a target is technically feasible before expanding into larger discovery campaigns.
How Do PROTAC Drugs Work Differently?
PROTAC drugs are heterobifunctional molecules designed to bring a target protein and an E3 ligase into proximity, enabling ubiquitin-proteasome-mediated degradation rather than simple functional inhibition. This event-driven mechanism can be attractive when sustained target removal may offer advantages over occupancy-based inhibition. BOC Sciences supports PROTAC discovery with services such as target ligand selection, E3 ligase pairing, linker planning, and structure-guided optimization to help teams build rational degrader libraries.
What Challenges Limit PROTAC Drug Development?
Key PROTAC development challenges include identifying a suitable target ligand, choosing the right E3 ligase recruiter, optimizing linker length and attachment sites, improving ternary complex productivity, and managing physicochemical properties associated with larger bifunctional molecules. These variables often interact, so small design changes can strongly affect degradation potency and selectivity. BOC Sciences helps address these issues through PROTAC design, linker optimization, target protein services, assay support, and iterative molecule refinement.
How Can Companies Start PROTAC Discovery Programs?
Companies usually begin PROTAC discovery by confirming target rationale, selecting a high-quality target-binding ligand, evaluating possible E3 ligase recruiters, and designing a focused set of linker variants for early degradation testing. A practical entry strategy is to combine computational design, chemical synthesis, and functional degradation assays in a coordinated workflow. BOC Sciences can support this process with integrated PROTAC design, synthesis-oriented planning, E3 ligand and linker resources, and research-focused degrader optimization services.
Explore More
Discover More Research Products
Explore featured products that can expand your research options and accelerate your next discovery.
Expert Services to Move Your Project Forward
Access end-to-end service solutions that help bring efficiency, flexibility, and expertise to your research pipeline.
News
Technical Information

