On the modern PROTAC landscape, aromatic linkers have been rediscovered as precision tools for fixing the POI–ligase pair into a catalytically ready conformation. In addition to a quantifiable reduction in conformational entropy brought about by replacing flexible alkyl or ethylene-glycol chains with planar rings, medicinal chemists can take advantage of newly emerged delocalized π-electrons that can engage in π-stacking interactions with histidine or phenylalanine side chains on either protein interface. The resulting ternary complex benefits from an additional "stickiness", often measurable as an increased residence time, allowing for lower systemic exposure without compromising on potency of degradation. As the aromatic core is synthetically orthogonal to most ligand exit vectors, it can be introduced without re-optimizing protecting-group strategies, and so rigidification is a low-risk late-stage manoeuvre rather than a discovery-stage gamble.
Introduction to Aromatic Linkers
One of the responses to the deleterious effect of excessive linker flexibility was the introduction of aromaticity. A six-membered ring on its own suffers from limitations in entropic penalties, susceptibility to metabolism, and uncontrolled binding geometries of its own. However, the aromatic pharmacophore conceptually represents a unique opportunity: a low-energy scaffold, by default planar, and whose rotational freedom is inherently constrained. The reduced number of rotatable bonds that sp2-hybridization affords effectively minimizes the entropic penalty of adopting the bioactive conformation and its improved membrane permeability is facilitated by the reduction of molecular ovality. Furthermore, aromaticity in its own right provides an apolar and chemically inert surface that, through π-stacking, can participate in protein–protein interactions with histidine, tyrosine, phenylalanine, or tryptophan residues that often decorate the POI–E3 interface. Individually weak but cumulative when multiple aromatic rings are stacked appropriately, this type of binding interactions can play the role of sewing the incipient ternary complex together. This conceptual breakthrough was validated in the first proof-of-concept examples when a 6-carbon alkyl chain was swapped out for a 1,4-phenylene scaffold to produce a significantly left-shifted degradation curve despite comparable binary affinity. This also functionally supported the non-scaffold hypothesis, and for reasons touched on above, derivatives followed rapidly with heteroaromatic replacements (pyridine, pyrimidine, thiophene, indazole, etc.) to tune basicity, dipole moment, and hydrogen-bonding potential while retaining planarity. The amenability of substitution in the ortho, meta, or para positions of such motifs also has been used to exert vectorial control on the exit pathway of each ligand, itself now believed to be a necessary condition for positive cooperativity.
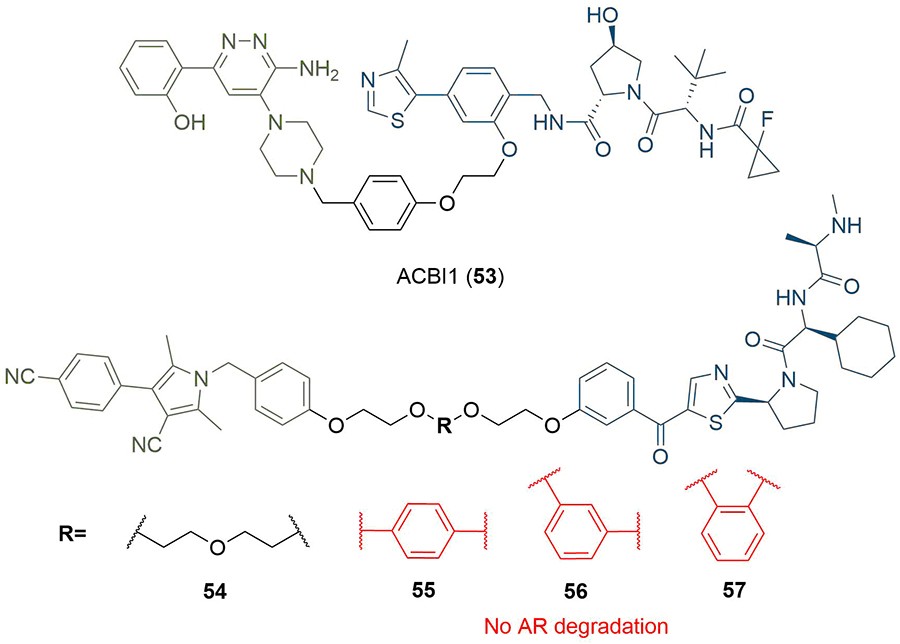 Fig. 1 PROTACs with aromatic linkers.1,2
Fig. 1 PROTACs with aromatic linkers.1,2
Structural Characteristics and Benefits
The performance of aromatic linkers can be understood by several properties. The planar π-system pre-defines a single vector between attachment points pre-organizing the PROTAC into an extended, rod-like shape and reducing steric repulsion as the POI and E3 surfaces come together. The delocalized electrons spread a partial charge across the ring creating quadrupole moments which can complement matching electrostatic patterns on the protein surface to increase binding energy without a formal charge. The thin profile of an aromatic ring (~50% the volume of a saturated 6-membered ring) reduces the linker's contribution to the often tight cleft formed at the ternary interface leaving more space for adaptive side-chain rearrangements. Aromatic C–H bonds have lower susceptibility to hydrogen-abstraction by cytochrome P450 enzymes relative to aliphatic C–H bonds, leading to better microsomal stability and slower clearance. The synthetic orthogonality of aryl halides, boronic acids, and terminal alkynes permits convergent assembly enabling medicinal chemists to rapidly scan regioisomers and heteroatom patterns. Collectively, this has resulted in a linker that is more than a scaffold to bridge the gap between ligands but actively contributes to molecular recognition, increased metabolic robustness, and efficient synthetic optimization. However, there is a delicate balance of lipophilicity to be managed. Stacking of sequential π-systems in solution can induce aggregation and false-positive biochemical read-outs. The inclusion of a single nitrogen in the ring or a solubilizing morpholine side-chain typically rebalances the physicochemical properties without interfering with the rigidifying effect.
Why Rigidity Matters in PROTACs?
The rigidity of a PROTAC affects all phases of the degradation process, from membrane permeation to ubiquitin transfer. A flexible linker samples many extended and collapsed states, only a subset of which orient the POI and E3 in an arrangement where they can bind simultaneously. The resulting entropic cost is directly reflected by shallow dose–response curves and submaximal degradation. Limiting the available torsional degrees of freedom biases the conformational distribution toward the productive conformation, prepaying the entropic price tag at synthesis instead of binding. The kinetic effect is accelerated association and decelerated dissociation of the ternary complex, which in turn makes it more likely that lysines on the POI will sample the E2–ubiquitin conjugate. Structural studies have also shown that many E3 ligases have a narrow groove between their substrate-recognition domain and the catalytic core; a rigid, linear linker spans this groove with no steric penalty, while a flexible chain kinks and sticks out, destabilizing the interface. Rigidity can also protect a PROTAC from metabolism: cyclic or aromatic moieties are devoid of the methylene groups that are the P450 enzyme's favorite oxidation sites, which prolongs the plasma half-life and reduces the necessary dose. On the physicochemical side, lower flexibility reduces the polar surface area and number of rotatable bonds, two properties that are well correlated with permeability and oral availability. On the other hand, too much rigidity can pass the point of diminishing returns, leading to a molecule that is too lipophilic or that orients the two proteins in an electrostatically unfavorable way; consequently, rigidity needs to be "titrated" to an optimal value, which is often achieved by "half-rigidifying" a PROTAC with one aromatic unit and a short, polar linker to provide a small amount of adaptive motion. In short, rigidity is not just a structural "nice to have", it is a kinetic and thermodynamic requirement for converting high-affinity binding into productive, sustained degradation.
Advantages of Aromatic Linkers
Aryl linkers have undergone an unsung evolution from simple scaffolders to gatekeepers of the PROTAC strategy. There is a sweet spot of properties they access rarely seen in other chemistries: a flat π-surface for dispersive interactions with a language understood by proteins, a preorganized backbone that is stiff and non-rotatable, and an electronic stability that is oxidative stress resistant. In contrast to pliable poly methylene chains, the aryl ring reduces the entropic cost of ternary-complex formation. The same delocalized orbitals also enable edge-to-face or off-set stacking with histidine and phenylalanine commonly lining the target–ligase interface, resulting in not only stronger binding but also a kinetic stabilization that affords the ubiquitin machinery time to work. Finally, the robust synthetic modularity of Suzuki or Sonogashira couplings makes the linker a canvas where electronics, dipoles and solubility profiles can be quickly redesigned, shortening the iterative cycles of design. In short, aryl linkers achieve many layers of optimization in rigidity, recognition, stability and synthetic agility, all in a small and light package that flexible chains have yet to match.
Enhanced π–π Interactions with Protein Surfaces
Few protein surfaces are smooth, and most are pitted with ridges of tyrosine, tryptophan, phenylalanine and histidine whose π-electron density is tantalizingly just above the peptide backbone. The aromatic linker offers a geometrically compatible analogue to these residues, such that the quadrupole moments of two facing rings can couple across the solvent gap. Subtle though the interaction is (often written off as "weak" in isolation), within the nanometer-scale gap of a nascent ternary complex it acts as a stitching force to hold the complex from falling apart prematurely. Edge-to-face geometries dominate when the linker ring is electron-poor; offset stacking prevails when both surfaces have complementary electron density; in either mode, the aromatic contact restrains the translational freedom of the POI relative to the E3 ligase, and thereby narrows the ensemble of productive poses. Because the aromatic cloud is delocalized, the contact is also forgiving of small structural drift, and provides a dampening buffer against the natural breathing of proteins. Critically, these contacts are orthogonal to hydrogen bonding and salt bridges, so they additively layer on top of existing polar interactions without steric conflict. The net result is a gentle but persistent adhesive force that lengthens the residence time of the target within the catalytic neighborhood, increasing the probability that a lysine side chain will meet the charged ubiquitin thioester. Unlike hydrophobic collapse, π–π stacking is directional; the linker does not therefore induce promiscuous aggregation, but rather reinforces the orientation selected by the warhead and the ligase ligand. By threading one or two carefully substituted aryl units into the linker, designers can therefore convert a transient encounter into a locked-in complex capable of sustained turnover.
Reduced Entropic Penalty in Ternary Complexes
Flexible linkers pay an entropy tax every time they freeze in a single productive conformation: the rotational and translational freedom that is lost translates directly into an entropic penalty that can erode binding affinity. Aromatic rings prepay this tax during chemical synthesis by locking contiguous atoms into a planar, low-energy array. When the PROTAC diffuses into the cell, it already carries a significant portion of the "binding cost" in its folded state, so the additional loss of freedom upon ternary-complex formation is modest. The pre-organized geometry also shrinks the search volume that the warhead and ligase ligand must explore, accelerating on-rates and reducing the likelihood of kinetically trapped, non-productive encounters. Molecular dynamics snapshots reveal that aryl-containing linkers visit far fewer conformational clusters than their alkyl counterparts, and the dominant cluster already resembles the bound pose; in contrast, flexible tethers wander through a broad Ramachandran-like space until a rare fluctuation aligns both ligands. From a thermodynamic standpoint, the reduced entropic cost manifests as a more favorable binding signature that is less sensitive to temperature elevation, an attribute that preserves activity under febrile or metabolically hot microenvironments such as inflamed tissue. Moreover, because the linker itself is no longer the main source of disorder, cooperativity becomes easier to detect and to optimize; small changes in warhead or ligase ligand are transmitted through the stiff connector without being smeared out by linker reconfiguration. Consequently, aromatic rigidity transforms the linker from an entropic liability into a thermodynamic capacitor that stores and relays structural information.
Improved Stability vs. Flexible Linkers
Metabolic and conformational stability are rarely mentioned in the same breath, but aromatic linkers offer both. The lack of benzylic or aliphatic C–H bonds remove the hotspots of microsomal oxidation; aryl rings can be oxidised only after an energetically uphill hydroxylation, and even these phenols are less susceptible to β-elimination than alkyl radicals. This results in slower clearance and less reactive electrophilic intermediates that could haptenize proteins and cause idiosyncratic toxicities. The stiff backbone is less likely to be sheared apart by the slingshot forces of crowded cytosolic proteins, which diminishes the chances that the warhead will be levered out of its binding pocket. Flexural rigidity also mitigates intramolecular cyclisation reactions (lactam or cyclic carbamate formation, for example) that can happen when nucleophilic side chains on the linker are forced into close contact by random coil collapse. When stored as a formulation, aromatic linkers are less likely to engage in auto-oxidative cross-linking, a failure mode that can befall PEGylated chains and lead to aggregation under light or heat stress. Photostability is also enhanced: the delocalized π-system quenches UV energy by rapid internal conversion, whereas flexible linkers can fragment by Norrish pathways. Finally, lower lipophilicity per unit length of an aryl ring than an alkyl chain of the same span means that partitioning into membranes stays modest, thus avoiding the micellar entrapment that speeds hydrolytic degradation. Taken together, these effects provide the PROTAC with a longer intracellular half-life, steadier exposure and less chance of off-target metabolites, all without resorting to exotic protective groups or pro-drug manoeuvres.
Popular Aromatic Linker Types
Of the increasing diversity of aromatic linkers, phenyl, biphenyl and heteroaryl motifs have been most widely used as backbone architectures. They represent a happy medium between synthesis-friendly and functional in that phenyl moieties offer a simple, flat rod that can be extended unidirectionally in small increments by homo-coupling to biphenyl or hetero-coupling to pyridine, thiophene or pyrazole. Each extension provides unique electronic, dipole and solubility properties without needing to re-synthesize the whole molecule. The resulting fragments slip more easily through the tight gaps that often separate target and ligase proteins, furnishing π-surfaces for transient π-stacking interactions, while also being much more resistant to oxidative degradation than alkyl linkers. Critically, all of these cores are available off the shelf as halogenated or boronated starting materials, so medicinal chemists can rapidly iterate substitution patterns rather than design de novo scaffolds. As a result, phenyl, biphenyl and their heteroaromatic analogs can serve as an interchangeable toolkit to expedite iteration cycles and reduce timelines from biochemical hits to cell degraders.
Phenyl and Biphenyl Linkers
The phenyl ring is probably the smallest rigid unit that still has a fully delocalized π-cloud, and that fact enables the phenyl moiety to serve as both spacer and recognition element in the same 6-C footprint. Because rotation about the two attachment vectors is governed only by relatively weak steric effects, a single phenyl segment can pivot a few degrees to compensate for the often-present small-angle twist between the warhead exit vector and the ligase ligand entry vector, which obviates the need for long, entropy-rich alkyl buffers. If the distance budget needs to be increased, a second phenyl can be cross-coupled to the first via a Suzuki or Negishi reaction to form a biphenyl rod whose length is predictably double that of the monomer but whose rotational freedom is far less than that of an eight-carbon alkyl chain. The inter-ring torsion can be biased toward planarity via ortho-substitution with small fluorine or methoxy groups, or conversely forced in the orthogonal orientation if steric clash with proximal protein side-chains is foreseen. From a metabolic perspective, the lack of benzylic C–H bonds means that microsomal oxidation must first cleave the aromatic π-system, a high-energy process that retards clearance and reduces the formation of reactive quinone-imines. Electron-donating or withdrawing substituents can be placed at the meta or para position to modulate dipole moment and thus fine-tune solubility without stretching the conjugated system too far, a move that is synthetically trivial because the required bromo- or borophenylalanine surrogates are shelf-stable. In short, phenyl and biphenyl linkers offer a length-scalable, electronically tunable and metabolically robust backbone that can be assembled in two or three steps from commodity reagents, making them the default first stop for most linker optimization campaigns.
High-Purity Phenyl Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity Phenyl linkers optimized for targeted protein degradation.
Pyridyl and Heteroaryl Variants
Replacing one or more of the carbons of a phenyl ring with nitrogen, oxygen or sulfur turns the neutral spacer into a heteroaromatic chameleon whose hydrogen-bonding propensity, basicity and dipole orientation can be dialed in with almost at will. Pyridyl rings, by far the most frequently deployed, present a lone pair that can either accept a proton or engage in weak hydrogen bonding with backbone NH groups that are often found at the periphery of the target–ligase interface; this subtle tug can tilt the entire PROTAC by a few degrees, steering the warhead deeper into its pocket without redesigning the exit vector. Regioisomerism offers an additional lever: a 3-pyridyl nitrogen projects its lone pair away from the linker axis, minimizing inductive withdrawal from the conjugated path, whereas 4-pyridyl placement aligns the dipole moment with the linker axis, useful when the ternary complex requires a small electrostatic zipper. Thiophene and furan insert a polarizable sulfur or oxygen that participates in dispersive contacts with methionine or cysteine side chains, while pyrazole or imidazole introduce an NH donor that can bridge to carbonyl oxygens, effectively stapling the chimera to the protein surface for a few extra milliseconds. Crucially, all of these heteroaryl halides and boronic acids are commercially available, so the same Suzuki couplings that work for biphenyls translate seamlessly to pyridyl-thiophene, pyridyl-pyrazole or even thiophene-furan hybrids, giving rise to a combinatorial matrix that can be explored in parallel. The end result is a linker that does not just span physical distance but also speaks the chemical dialect of proteins, using hydrogen bonds, dipole alignment and transient π-stacking to hold the nascent ternary complex together long enough for ubiquitin to arrive.
Case Studies in Literature
In recent medicinal-chemistry literature, rigid aromatic linkers have been used repeatedly to turn modest biochemical hits into cell-active degraders. In the same studies, however, a series of subtle liabilities have also been identified that leave much to be desired. Single-ring phenylene or biaryl rods have enabled picomolar degradation of kinases, epigenetic readers and helicases with one or two synthesis iterations, often outperforming flexible analogues that required longer synthesis and higher dose. On the other hand, crystallographic and cellular data suggest that an overly π-rich strand can stack with off-targets, collapse in polar media or elongate the molecule past a narrow "sweet spot" for oral uptake. The following sections highlight representative success stories before dissecting the boundary conditions that must be considered if the aromatic advantage is to be preserved.
Success Examples with Rigid Aromatic Linkers
One of the first proof-of-principle efforts involved substituting a four-carbon alkyl chain with a single para-phenylene group in a PROTAC targeting a DNA-repair helicase. The aromatic bridge brought the binding sites closer together, pre-organized the exit vectors, and established an edge-to-face interaction with a tyrosine residue sandwiched between the ATP-binding site and the surface of the recruited E3 ligase. After two iterations (introduction of a meta-fluoro substituent and swapping of the exit vector amide to an ether), the degrader exhibited complete target clearance at sub-nanomolar exposure while the unbridged, flexible parent remained inactive even at micromolar concentrations. In another report, a bromodomain reader protein was targeted: in this case, a biphenyl rod was locked near coplanarity by an ortho-methoxy substituent, which in turn introduced a twist that reproduced the dihedral angle between the acetyl-lysine pocket and the von Hippel–Lindau recognition site. X-ray crystallography revealed a sandwich π-stack in which the central ring of the linker made contact with an exposed histidine of the bromodomain and a phenylalanine of the ligase, a binding mode not seen with the alkyl comparator. Cellular data demonstrated prolonged degradation lasting over 24 h despite wash-out, which is consistent with slow dissociation from the ternary complex. Perhaps the most illustrative example to date was found for a pyridyl-thiophene hybrid inserted into a STAT3-directed PROTAC: the nitrogen lone pair accepted a hydrogen bond from a backbone NH at the rim of the SH2 pocket, which tilted the entire chimera by roughly twelve degrees and swung a peripheral lysine residue into the catalytic cleft. The aromatic hetero combo restored activity against a mutant isoform that had evolved resistance to conventional flexible degraders, demonstrating how directional contacts can rescue otherwise futile architectures. In these different cases, rigidity was not pursued as an end in itself, but as a conduit for creating new, weak yet specific contacts that stitched the nascent complex together long enough for poly-ubiquitin transfer to occur.
Limitations and Considerations
For each success story in the literature, there is an equal train of aryl-rich mimics that were trapped at the biochemical barrier or failed in vivo. The most common pitfall is π-stacking promiscuity: a long poly-aryl rod can fit into the ATP binding site of kinases, the RNA groove of helicases or the hydrophobic slit of nuclear receptors, resulting in false-positive degradation signals that disappear after the linker is trimmed. Crystal structures of such off-target complexes frequently display the aromatic plane sandwiched between two phenylalanines, a geometry that is aesthetically satisfying but pharmacologically disastrous. Solubility issues form a second, more insidious barrier; consecutive rings increase the crystal packing energy and decrease the kinetic solubility below the threshold required for reliable cell permeation, resulting in flat dose-response curves that resemble target resistance. Metabolic stability, which is often much better than aliphatic chains, can flip to the opposite extreme when electron-donating methoxy or methylthio groups are introduced to modulate electronics; these moieties are then metabolized by cytochrome-mediated demethylation, releasing catechol or thiophenol moieties that haptenize proteins and lead to clearance. Finally, linker length rigidity can exceed the optimum, forcing the POI and E3 ligase into an electrostatically repulsive conformation that offsets any enthalpic benefit from π-contacts. Several publications report "hook effect" profiles in which linker elongation initially improves but then abolishes degradation, with the point of inflection coinciding with the loss of positive cooperativity. Taken together, the literature suggests a cautious titration of aromatic density, counteracted by polar spacers or strategic heteroatoms, to ensure that the benefits of rigidity and recognition are not negated by solubility, promiscuity or metabolic red flags.
Ordering Options for Aromatic Linkers
At BOC Sciences, we offer a curated selection of aromatic linkers specifically designed for PROTAC development. Each linker is synthesized under strict quality control standards to ensure high structural integrity, rigidity, and reproducibility - all essential for optimizing ternary complex formation and π–π stacking interactions. Whether you're in the early stages of lead identification or fine-tuning linker geometry for advanced candidates, our ready-to-ship inventory and custom synthesis services provide the flexibility and quality your research requires.
Stock Availability and Functional Handles
We maintain an extensive catalog of aromatic linkers readily available for immediate dispatch, including:
- Phenyl, biphenyl, pyridyl, and heteroaryl scaffolds optimized for rigidity.
- Pre-functionalized variants featuring reactive handles such as bromo, iodo, boronic acid (Bpin), azide, and alkyne groups - ideal for fast conjugation or coupling chemistry.
- High purity levels, confirmed by HPLC, LC-MS, and NMR analysis, with full Certificate of Analysis (COA) documentation.
Our in-stock products are packaged for stability. Researchers can rely on our consistent quality and analytical transparency to accelerate synthesis and scale-up without supply interruptions.
Custom Synthesis for Research Projects
If your PROTAC design requires a specific aromatic framework or unique linker configuration, our custom synthesis program provides full flexibility. We can modify:
- Core scaffold type (mono-, bi-, or tri-aryl systems).
- Substitution pattern and orientation (para-, meta-, ortho- connectivity).
- End-group functionalization to match your E3 ligase or target ligand chemistry.
- Electronic tuning through heteroatom incorporation or ring fusion.
Our chemists collaborate directly with your R&D team to ensure the resulting linker aligns with your structural, solubility, and permeability goals.
Partner with Us for High-Purity Aromatic Linkers
Aromatic linkers play a pivotal role in PROTAC design, offering the rigidity and π–π interaction potential needed to stabilize ternary complexes and enhance binding specificity. Selecting a high-quality, well-characterized aromatic linker can make the difference between marginal and breakthrough activity. At BOC Sciences, we supply high-purity aromatic linkers trusted by drug discovery teams worldwide. From off-the-shelf phenyl and pyridyl linkers to fully customized aromatic scaffolds, we deliver precision chemistry backed by analytical reliability and rapid global shipping.
Contact our technical specialists today to request a quote, explore our inventory, or discuss a custom aromatic linker synthesis for your next PROTAC development project. Let's build the rigid linkers that drive your discovery forward - with chemistry you can count on.
FAQs
1. Why are aromatic linkers important in PROTAC design?
They introduce rigidity and facilitate π–π stacking, improving ternary complex stability.
2. Which aromatic scaffolds are available?
We offer phenyl, biphenyl, pyridyl, and heteroaryl linkers in multiple substitution patterns.
3. Can aromatic linkers reduce flexibility too much?
Overly rigid structures can limit binding adaptability, so optimal balance is key.
4. Do you provide functionalized aromatic linkers?
Yes, pre-functionalized variants (bromo, azide, alkyne, Bpin) are available for rapid coupling.
References
- Image retrieved from Figure 1 " PROTACs with aromatic linkers," Troup R I.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Troup R I, Fallan C, Baud M G J. Current strategies for the design of PROTAC linkers: a critical review[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 273. https://doi.org/10.37349/etat.2020.00018.
- Cieślak M, Słowianek M. Cereblon-recruiting PROTACs: Will new drugs have to face old challenges?[J]. Pharmaceutics, 2023, 15(3): 812. https://doi.org/10.3390/pharmaceutics15030812.
- Zografou-Barredo N A, Hallatt A J, Goujon-Ricci J, et al. A beginner's guide to current synthetic linker strategies towards VHL-recruiting PROTACs[J]. Bioorganic & Medicinal Chemistry, 2023, 88: 117334. https://doi.org/10.1016/j.bmc.2023.117334.
- Nassar H, Sarnow A C, Celik I, et al. Ternary Complex Modeling, Induced Fit Docking and Molecular Dynamics Simulations as a Successful Approach for the Design of VHL‐Mediated PROTACs Targeting the Kinase FLT3[J]. Archiv der Pharmazie, 2025, 358(4): e3126. https://doi.org/10.1002/ardp.202500102.
