A consistent result across crystallographic and live-cell datasets has been that a simple substitution of a phenyl with a pyridyl ring buried inside the PROTAC backbone is a surprisingly powerful tuning knob for ternary-complex half-life. The additional nitrogen provides three simultaneous benefits: a hydrogen-bond acceptor that can ligate ordered water at the PPI interface, an increase in the electron-deficiency of the π-stack so that edge-to-face contacts with histidine or phenylalanine side chains become more enthalpically favorable, and a lower barrier to late-stage diversification as the lone pair tolerates electrophilic bromination or nucleophilic substitution without loss of aromaticity. The phenyl-to-pyridyl switch is therefore now viewed less as a minor isosteric edit and more as a conformational lock that can rescue programmes once flexible linkers have hit their polarity ceiling.
Introduction
Rigid linkers were originally mechanical spacers used to maintain a physical distance between the warhead and E3 ligand, but have since become allosteric determinants that control whether the ternary complex is long-lived enough to be ubiquitylated for degradation. Fixing rotatable bonds into aryl/heteroaryl planes pre-pays the entropic penalty for binding, reduces the number of accessible conformations and forms geometrically constrained π-surfaces that can sandwich against either protein partner. The sections below will examine how this conformational restriction impacts cooperativity and why the decision to use phenyl vs pyridyl can affect on-rate and residence time without changing linker length.
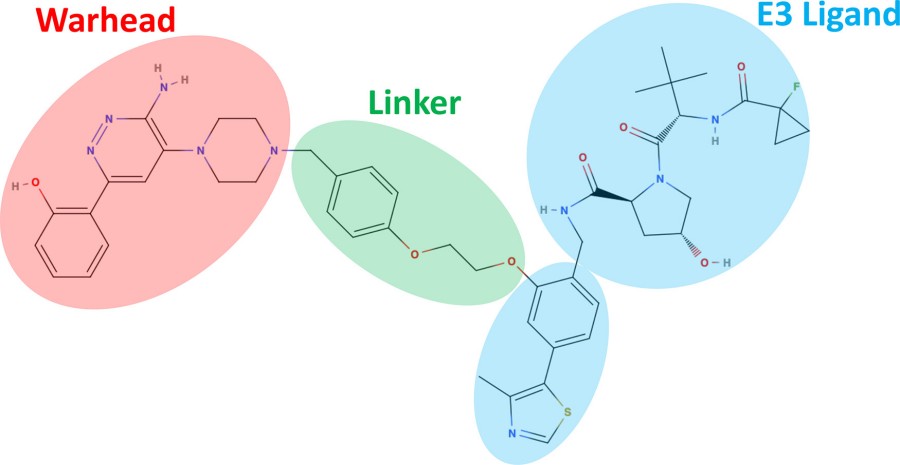 Fig. 1 Example of a heterobifunctional degrader molecule.1,2
Fig. 1 Example of a heterobifunctional degrader molecule.1,2
Flexibility comes at a steep thermodynamic cost when two proteins must be bridged in one productive pose. A floppy alkyl chain populates hundreds of extended, collapsed and looped conformers. Only a tiny fraction juxtaposes the catalytic lysines of the POI and the ubiquitin-loaded E2–E3 machinery, so the observed affinity is watered down by a punishing entropic penalty. Rigid segments, in contrast, arrive pre-organized. The low-energy conformer already resembles the bound state, so the cost of translation/rotation is paid during synthesis, not at the binding site. This entropic pre-payment appears as faster association, slower dissociation and a narrower thermal window over which activity is lost. Rigidity also shrinks the spatial search volume, forcing the warhead and ligase ligand to find their respective pockets in one diffusional encounter rather than after repeated bind–release cycles. Mechanically, an aryl rod acts like a molecular yard-stick: its length is fixed within a few tenths of an ångström, so designers can lengthen or shorten the span in predictable increments until the lysine side chain is deposited within reach of the E2 active site. Equally important, the planar π-cloud can participate in edge-to-face or offset stacking with phenylalanine, tyrosine or histidine lining the inter-protein cleft, providing enthalpic glue that supplements the binary affinities of the individual ligands. Finally, rigid linkers protect the molecule from its own metabolic fragility: the absence of freely rotating methylene groups removes the primary sites of cytochrome P450 oxidation, extending intracellular lifetime and allowing lower exposure. Taken together, rigidity acts as a thermodynamic capacitor, kinetic accelerator and metabolic shield, three attributes that explain why the first reflex of most optimization campaigns is to trade floppy carbon chains for aromatic planes.
Phenyl vs. Pyridyl as Linker Choices
Phenyl and pyridyl differ by only a single atom, but the difference of that nitrogen substitution echoes through conjugation, solubility and recognition in ways that can make or break a degrader. Phenyl provides a symmetrical, non-polar slab whose quadrupole moment is null; it slides easily between protein aryl side chains without a strong directional preference, a neutrality that can be an asset when the binding groove is narrow and apolar but which can also encourage off-target π-stacking with ATP sites or lipid bilayers, and does nothing to offset the rising logP that comes with each added ring. Pyridyl disrupts that symmetry: the nitrogen pulls electron density, creating a molecular dipole that can be pointed at backbone amides or organized water molecules in the protein periphery. The lone pair is a weak hydrogen-bond acceptor, and can frequently be used to form a water-bridged contact to a backbone NH that is too far away for direct H-bonding; this transient bridge can tack the linker to the protein surface, increasing residence time without increasing molecular weight. The regiochemistry is an additional dial: 3-pyridyl keeps the group conjugated but moves the lone pair far from the linker axis, reducing inductive withdrawal, while 4-pyridyl points the dipole moment along the rod, which can be helpful when the ternary interface has space for a small electrostatic zipper. From a solubility standpoint, the nitrogen lowers melting point and provides a weakly basic center that can be protonated at lysosomal pH, improving aqueous handling without the steric bulk of a pegylated chain. Metabolically, the heteroatom discourages direct hydroxylation and can be oxidized to N-oxide, a metabolite that is often more polar and more readily excreted than the corresponding phenol from a phenyl linker.
Phenyl Linkers
Phenyl rings are synthetic versatility, predictable metabolic stability, and conformational rigidity that have made them some of the most commonly used rigid cores in degrader discovery. The six-fold symmetry provides multiple orthogonal exit vectors without the addition of stereogenic centers, and the electron-rich π-cloud is capable of edge-to-face interactions that can impart hidden enthalpy to the ternary complex. However, the hydrophobic surface area that can facilitate membrane permeation can also promote off-target aggregation or plasma-protein binding if the hydrocarbon chain length is not carefully tuned. The following sections will explore these trade-offs and provide a checklist for when a simple phenyl bridge may be an asset or a liability.
Benefits of Phenyl in Stability and Availability
The primary benefit of the phenyl ring, however, is synthetic accessibility: With several hundred boronic acids, bromides and iodides available off-the-shelf at commodity cost, many groups can quickly build bi-aryl linkers from scratch using Suzuki or Sonogashira coupling methods without investing time in creating a custom synthesis. After attachment, the extended π-system also avoids metabolic traps like oxidative cleavage because P450 first needs to invest energy into epoxidizing an electron-rich alkene; in practice the ring remains intact long enough for the degrader to bind to multiple target copies in vivo, which in turn allows for lower clinical doses. The planar scaffold also pre-organizes the PROTAC into a structure that closely resembles the bound state, reducing the entropic cost that flexible tethers must overcome when the POI and E3 ligase eventually meet. An analytical benefit of the UV chromophore at 254 nm, meanwhile, is an immediate handle for quantifying reaction conversion and final product purity that accelerates batch release without the need for a mass-tag derivatization. Finally, I mention in passing that regulatory toxicologists are already very familiar with biphenyl fragments from many years of agrochemical and polymer science, so the safety discussion rarely lingers over residual solvent or metal content once ICH guidelines are satisfied.
Limitations of Simple Aromatic Structures
The same π-rich surface that accelerates membrane transit can also nucleate π–π stacking between PROTACs, leading to colloidal precipitates at high concentration or upon freeze–thaw cycling. The amount of time a compound spends in the unbound state increases slowly with ring count, so adding a single additional phenyl can reduce the unbound fraction by half and push the effective dose outside the solubility envelope. Metabolic stability, though often excellent, is correspondingly variable once two or more rings are fused: for example, sequential hydroxylation can produce para-quinone mimics that deplete glutathione and set off idiosyncratic alarms. Perhaps most important, lack of a directional hydrogen-bond acceptor means that water molecules bridging POI and E3 have to be recruited from the protein itself; if mutations increase the size of the interfacial gap, the phenyl linker cannot compensate by forming new H-bonds, a rigidity that can break cooperativity. These factors strongly suggest that simple phenyl bridges are best used for targets whose crystal structure already exposes a hydrophobic corridor between ligand-binding sites and where the therapeutic window can accommodate modest increases in lipophilicity.
High-Purity Phenyl Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity Phenyl linkers optimized for targeted protein degradation.
Pyridyl Linkers
Pyridyl linkers strike a middle ground between hydrophobic phenyl rods and flexible polar tethers, by contributing a lone-pair-bearing nitrogen that can both accept hydrogen bonds and rotate the molecular dipole without significant added mass. In several recent degradation campaigns, the same pyridine that stiffens the backbone has also served as a chemical gyroscope: it tilts the whole chimera a few degrees so that catalytic lysines face the E2–E3 active site, while simultaneously providing a site of protonation that rescues aqueous solubility at acidic endosomal pH. The following sections explain how these electronic and vectorial benefits come about, and where the tradeoffs are.
Added Polarity and H-Bonding Potential
Beyond substituting a C–H with a lone pair, a nitrogen insertion also distorts the aromatic π-cloud and endows the molecule with a molecular dipole, which orients itself along other backbone amides or structured waters which commonly line the rim of the target–ligase interface. The pyridyl nitrogen acts as a relatively weak hydrogen-bond acceptor whose affinity can be modulated through regiochemistry: a 3-pyridyl substitution will deshield the para carbon but leaves the lone pair partially protected, while a 4-pyridyl position places the nitrogen itself directly along the linker axis and open to direct engagement by a water bridge or an NH donor. These dynamic hydrogen bonds are sufficiently weak as to not kinetically trap the complex but can still contribute a few milliseconds to residence time, which is an eternity in ubiquitination time. As the interaction is directional, it can also compensate for small exit-vector angle mismatches, help bury the warhead deeper into its pocket, and do so without having to redesign the core of the ligand. From a physicochemical perspective, the additional polarity decreases the melting point of the solid PROTAC and improves kinetic solubility in buffered media, making colloidal aggregation less likely in PROTACs than in purely carbocyclic linkers. At lysosomal pH the partial protonation of the nitrogen also creates a cationic handle that aids in membrane escape, but is sufficiently shielded at neutral cytosolic pH as to avoid becoming permanently charged. At the same time, the same lone pair can engage in π-back-donation with heme or copper centers, leading to occasional inhibition of cytochromes or metalloenzymes; it is therefore important to do control experiments monitoring off-target enzyme activity to ensure that the degradation signature is not being confounded by a binding event with a metalloprotein. The extent of this risk can be dialed back by carefully tuning the electronics (e.g., fluorination, remote methoxy substitution) without entirely forgoing the hydrogen-bonding dividend, making the pyridyl linker a chemically innocuous but pharmacologically critical element.
Improved Orientation in Ternary Complexes
In addition to polarity, the pyridine ring also acts as a geometric knob that reorients the entire chimera within the narrow cleft formed by target and E3 ligase. Crystal structures of SMARCA2–VHL complexes show that regioisomeric pyridyl linkers can tilt the ligase domain up to twelve degrees to reposition a surface lysine directly above the ubiquitin-loaded E2 active site. The alternate option—a symmetrical phenyl ring—whose quadrupole moment is too isotropic to bias rotational registration. The nitrogen-induced dipole produces a "compass needle" effect where the ring spontaneously orients with the negative end of the dipole facing electron-poor backbone regions and the positive end tucked against π-rich side chains, effectively stapling the nascent complex into a catalytically competent pose. Molecular dynamics trajectories indicate that pyridyl-linked PROTACs sample fewer rotational clusters and remain longer in the productive basin, which is translated into faster ubiquitin transfer and more complete degradation at low exposure. Critically, the orientation advantage is sensitive to substitution pattern: ortho-fluorination can lock the ring into a coplanar arrangement, whereas meta-methoxy substitution introduces a slight twist to compensate for dihedral mismatches at the protein interface. The kinetic consequence is a slower off-rate that outlives transient dips in intracellular concentration, giving the ubiquitin machinery time to poly-merise the substrate even under wash-out conditions. Together, these data place the pyridyl linker not just as a passive spacer but as a conformational helmsman that converts electronic subtlety into macroscopic degradation potency.
Comparative Analysis
Structure-activity comparisons made side by side have shown that exchanging one phenyl for one pyridyl, or reducing a biphenyl to a thiophene-pyridine chimera, can reverse the order of cell potency rankings without altering the binary binding. In several of the above series, rigid aromatics outpaced flexible alkanes in depth and duration of degradation, but their lead shrank when the target–ligase distance strayed from the linker's intrinsic length. The next few sections attempt to combine these trends into qualitative heuristics for when to consider a scaffold jump and which design knobs to focus on.
Performance Differences in SAR Studies
A variety of structure-activity interrogations also indicate that the single largest leaps in activity are typically achieved during the initial step in this chain-extension progression. Switching from a 4-6 carbon alkyl spacer to a phenylene or pyridylene unit often reduces the active window by an order of magnitude with marginal improvements thereafter, and the simplest interpretation of this pattern is that the initial gain is an entropically driven benefit of pre-organization, while further increases in length take the ternary complex beyond an ideal aspect ratio. Swapping one of these for a biphenyl or thiophene-pyridine pair often results in greater depth of action but comes with correspondingly reduced marginal returns. Regioisomeric swapping within a given heteroaryl collection can have as strong an impact on activity as does changing linker length: for example, shifting a pyridyl linker's nitrogen from the meta to the para position has the consequence of flipping the direction of the molecular dipole moment, with the result that a flat SAR can be converted to a strong sigmoid. Subtle changes in linker electronics also have important consequences, with electron-withdrawing F or Cl often having the effect of sharpening the degradation response by pulling electron density out of the π-cloud and thereby increasing edge-to-face interactions, while electron-donating methoxy substituents have the sometimes-desirable effect of widening the window of action but can also be susceptible to oxidative demethylation that reduces metabolic stability. Intriguingly, some series exhibit a U-shaped response to the degree of aromaticity: activity improves with the first two rings but then diminishes when a third aryl group is appended, and the rationale for this arc has been ascribed to crystal packing enthalpy and diminished kinetic solubility, rather than insufficient binding energy.
Decision Criteria for Switching
A scaffold swap is generally considered when at least two of the following warning signs are present: shallow dose–response slope, incomplete maximal degradation, or a cell activity loss that cannot be restored by enhancing the ligands' own permeability. When microsomal stability is insufficient and LC–MS traces show hydroxylation on benzylic methylenes, a jump from alkyl to aryl is often needed to eliminate the vulnerable C–H bonds. If crystallography or docking shows that the lysine to be ubiquitinated is situated just beyond the catalytic reach, but elongating the flexible linker only results in a plateauing of efficacy, then a rigid, fixed-length spacer like biphenyl or phenyl-pyridyl should be explored first. On the other hand, if degradation is strong but solubility or aggregation limits are reached, backtracking to a shorter heteroaryl ring or placing a single polar heteroatom (morpholine, sulfone) between two aryls can often recalibrate without losing the entropic bonus. Regiochemistry can be critical when the ternary model reveals the linker brushing up against a charged patch: a 4-pyridyl nitrogen can accept a hydrogen bond from an arginine guanidinium, while a 3-pyridyl orientation will not be sterically impeded and keep neutrality. Finally, metabolic liabilities can guide back-and-forth toggling: if previous series show that pyridine N-oxide formation speeds clearance, returning to a phenyl core and making up with a peripheral morpholine can preserve potency while shutting the oxidative gate. In summary, the switch is not based on a single criterion but on a weighted scorecard that intertwines geometric fit, electronic complementarity, solubility headroom and metabolic robustness; only when two or more parameters simultaneously fall short of their acceptance gates should the chemist give up the current scaffold and commit to a new aromatic dialect.
Buying Guide
At BOC Sciences, we provide high-quality phenyl and pyridyl linkers specifically engineered for PROTAC development and ternary complex stabilization. Our aromatic linker collection includes a wide range of rigid scaffolds, each verified for purity, structural integrity, and chemical reactivity to help you streamline synthesis and improve experimental reproducibility.
Pre-Functionalized Phenyl and Pyridyl Linkers
We stock a comprehensive selection of pre-functionalized phenyl and pyridyl linkers designed for efficient conjugation in PROTAC assembly. Available options include:
- Para-, meta-, and ortho-substituted phenyl linkers for precise vector orientation.
- Pyridyl linkers with built-in nitrogen heteroatoms that enhance hydrogen bonding and dipole interactions within ternary complexes.
- Reactive handles such as bromo, iodo, boronic acid (Bpin), azide, and alkyne groups for seamless coupling reactions.
- High purity, confirmed by HPLC, LC-MS, and NMR, with complete Certificate of Analysis (COA) for every batch.
All linkers are available in standard research quantities and packaged for stability under controlled conditions.
Bulk Supply and Custom Requests
For teams scaling from discovery to preclinical stages, we offer bulk supply and custom synthesis services for phenyl and pyridyl linkers. Our customization capabilities include:
- Adjusting ring substitution patterns to modify orientation and rigidity.
- Incorporating heteroatom substitutions or mixed aryl-heteroaryl frameworks for fine-tuned polarity.
- Delivering custom functional groups (amine, azide, alkyne, NHS, maleimide) based on your conjugation strategy.
- Producing GMP-adjacent or multi-kilogram batches for advanced R&D programs.
Our chemists work closely with clients to optimize linker geometry, ensuring compatibility with specific E3 ligase binders and protein targets. We provide quick turnaround, detailed documentation, and full analytical support for every order.
Partner with Us for Reliable Phenyl and Pyridyl Linkers
Rigid linkers such as phenyl and pyridyl scaffolds are central to the design of next-generation PROTACs, where structural orientation and electronic effects determine ternary complex stability and degradation efficiency. Choosing the right linker partner ensures both chemical precision and project success. At BOC Sciences, we specialize in high-purity phenyl and pyridyl linkers, combining analytical rigor, scalable synthesis, and global logistics to support your discovery goals.
Contact our technical team today to request a quote, discuss custom synthesis, or check stock availability for your desired linker. Accelerate your PROTAC research with rigid linkers built for stability, precision, and performance.
FAQs
1. What's the difference between phenyl and pyridyl linkers?
Pyridyl linkers contain nitrogen atoms that enhance polarity and orientation control compared to phenyl linkers.
2. When should I switch from phenyl to pyridyl?
When polarity, hydrogen bonding, or 3D alignment improvements are needed for complex stability.
3. Are these linkers compatible with common E3 ligases?
Yes, both phenyl and pyridyl linkers are compatible with VHL, CRBN, and IAP ligases.
4. Can I request custom aromatic designs?
Absolutely—custom pyridyl or mixed aryl–heteroaryl scaffolds can be synthesized upon request.
References
- Image retrieved from Figure 1 "Example of a heterobifunctional degrader molecule," Mostofian B.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Mostofian B, Martin H J, Razavi A, et al. Targeted protein degradation: advances, challenges, and prospects for computational methods[J]. Journal of Chemical Information and Modeling, 2023, 63(17): 5408-5432. https://doi.org/10.1021/acs.jcim.3c00603.
- Kelm J M, Pandey D S, Malin E, et al. PROTAC'ing oncoproteins: targeted protein degradation for cancer therapy[J]. Molecular Cancer, 2023, 22(1): 62. https://doi.org/10.1186/s12943-022-01707-5.
- Troup R I, Fallan C, Baud M G J. Current strategies for the design of PROTAC linkers: a critical review[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 273. https://doi.org/10.37349/etat.2020.00018.
