Alkyl linkers have seen a resurgence as a strategic alternative to ethylene-glycol tethers when the therapeutic objective is to optimize for passive diffusion across the lipid bilayer. Replacing the ether oxygens with methylene groups strips the spacer of hydrogen-bond acceptors, reduces topological polar surface area and allows the entire degrader to assume a more compact, cylindrical form factor in the membrane interior. The compromise in aqueous solubility and increased propensity for non-specific binding can be ameliorated by the inclusion of a single strategically-placed heteroatom or by truncating the chain to the shortest length necessary to span the POI and E3 exit vectors. Recent comparative permeability studies have shown that at matched lipophilicity, alkyl-linked degraders outperform PEGylated analogues in parallel artificial membrane assays, which correlates to improved unbound concentrations in plasma and brain tissue. As such, alkyl linkers are no longer considered legacy motifs of early tool compounds but are actively re-incorporated during lead optimization after the ternary complex geometry has been frozen and the main objective shifts to systemic exposure.
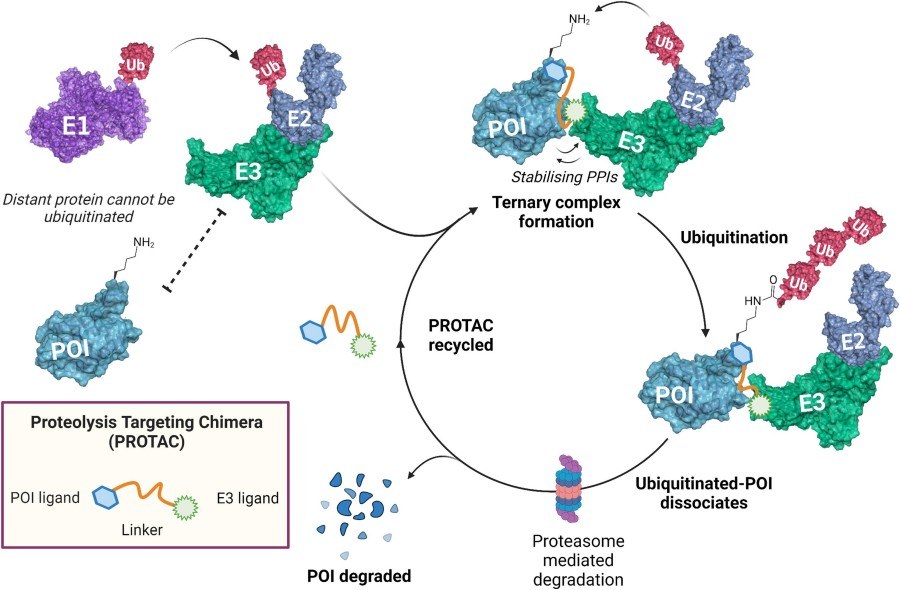 Fig. 1 Targeted protein degradation catalysed by a PROTAC.1,5
Fig. 1 Targeted protein degradation catalysed by a PROTAC.1,5
Introduction to Alkyl Linkers
Alkyl linkers are saturated or unsaturated hydrocarbon chains that bridge the two pharmacophores of a PROTAC without contributing recurring ether oxygens. Their main attraction is synthetic economy: a linear alkyl di-acid or amino-alcohol can be grown two carbons at a time with malonate chemistry or cross-metathesis, then "topped off" with conventional activating agents. Since the resultant spacer is lipophilic and metabolically "inert", it is rarely a site of oxidative cleavage, and the molecule is often fragmented at the ligand–linker amide or ester bond instead, making metabolite identification easier. The lack of repeating polar units also shortens the molecular weight addition per "virtual rotamer", which allows teams to span distance without crossing the size threshold that provokes biliary clearance. These properties have helped return alkyl linkers to favor for programmes that must penetrate the CNS quickly or require high oral absorption, so long as solubility/aggregation can be tamed with salt formation or nanoparticle delivery.
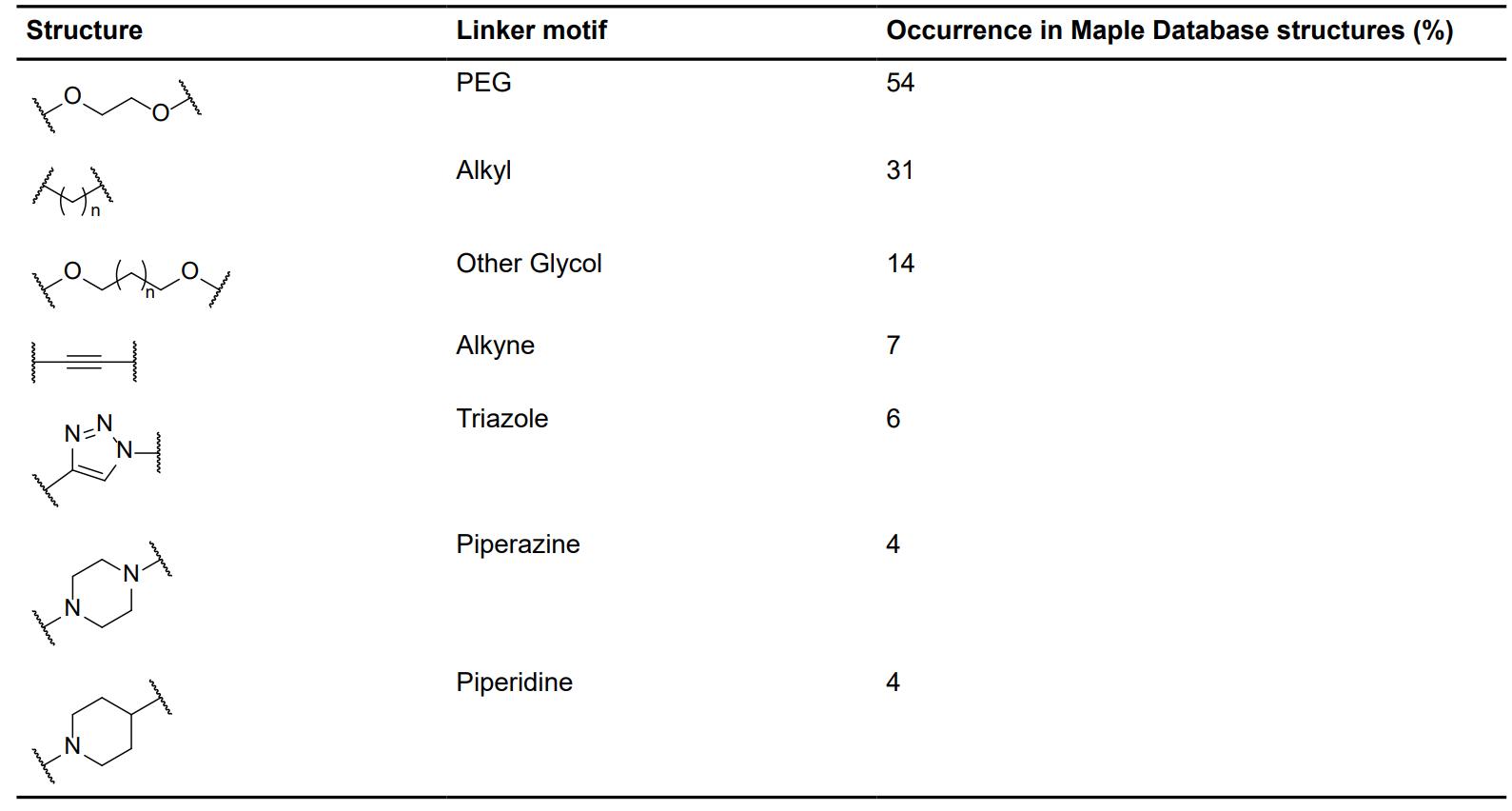 Fig. 2 Occurrence of selected linker motifs in the Maple database of published degrader structures.2,5
Fig. 2 Occurrence of selected linker motifs in the Maple database of published degrader structures.2,5
Structural Characteristics
Alkyl linkers can be viewed as conformational rheostats whose torsional potential is a function of steric repulsion, gauche interaction, and (optional) π-conjugated segments. Linear linkers access a distribution that spans end-to-end distances approaching the contour length, a geometry that generally maximizes the spacing between target and E3 surfaces and minimizes steric strain within the incipient ternary. A single methyl branch at the center shifts the distribution toward kinked geometries, contracting the span and subtly shifting exit vectors by ~1 bond length; such small changes have been used to flip cooperativity without changing the covalent chemistries of either warhead. Unsaturated moieties (single double bonds or polyenes) provide planar segments that limit torsion without being so long as to preclude induced fit. Heteroatom spacers, most often oxygen or nitrogen, impart local polarity that improves aqueous solubility without obviating the global hydrophobic character needed for membrane permeability; the ether or tertiary amine is also a metabolic liability that can be modulated by neighboring branches to adjust the rate of oxidation. Critically, repeating ether groups are not present, so peroxide-mediated chain cleavage that shortens PEG polymers in vivo does not occur, and thus exposure is driven by hydroxylation rather than chain scission, a mechanistic difference that often results in lower clearance and shallower dose–response profiles. Collectively, these design parameters endow chemists with angstrom-level control over spatial presentation while maintaining the synthetic accessibility of traditional organic fragments.
Differences from PEG Linkers
The key difference between PEG and alkyl linkers is how they achieve water solubility: the former via multiple presentation of ether oxygens' lone pairs, the latter via an imbalance in hydrophobic surface area and smaller polar handles. The latter strategy also has the distinct advantage of being lipid-like, which fits well with an oral medicinal chemistry mindset. This difference in design philosophy leads to experimentally testable hypotheses. For one, alkyl analogs have higher apparent permeability in PAMPA assays as there are fewer hydrophilic ethylene glycol units to pay a transfer penalty upon moving from aqueous donor phase to lipid acceptor phase. This improvement is especially true if the ligand has existing hydrogen bond donors (HBDOs) that would be sequestered into unfavorable intramolecular contacts in the PEG version. Metabolic stability would also shift from cleavage to oxidation: PEG chains become shorter progressively through ether peroxidation, while alkyl chains are typically metabolized at the terminal or sub-terminal position to hydroxylation which retains the overall chain length and degrader activity. Solubility is another differentiating aspect moving from entropy driven PEG hydration to enthalpy driven micelle like dispersion. In formulation, short alkyl linkers are often used together with ionizable counter ions, or with cyclodextrin carriers to obtain a similar drug exposure but at a reduced pill burden. Synthetic scalability is also more favorable for hydrocarbon chemistry (hydrogenation, alkylation, olefin cross-coupling, etc.) on standard pilot plant equipment with no anhydrous conditions needed, or expensive polyether protecting groups, leading to shorter campaign time and lower cost of goods. The lack of polydisperse reagents also gives a clearer impurity landscape from a regulatory standpoint: each carbon specific homologue can be isolated and treated as a discrete chemical entity, rather than the statistical tailing one often sees when setting specifications for PEG starting materials. For these reasons, alkyl and PEG linkers are complementary instead of competing toolkits in the design of degraders.
Benefits of Alkyl Linkers
The physicochemical signature of alkyl linkers can pull degraders past the polarity ceiling created by repeated ether oxygens. The elimination of hydrogen-bond acceptors and the provision of a hydrocarbon surface compress topological polar surface area, increase lipophilic efficiency and promote the overall structure to collapse into a membrane-friendly cylinder. The real-world pay-off is more than just an improved cLogP: the same modification will often double passive permeability, steepen the dose–response curve in whole-cell assays and increase oral exposure without resorting to the elaborate formulation maneuvers that longer PEG chains often require. In other words, alkyl spacers exchange aqueous generosity for membrane affinity, a trade that becomes crucial when the therapeutic target is on the far side of a tight epithelium or when rapid intracellular accumulation is needed to saturate efflux pumps.
Improved Membrane Permeability
Membrane permeation is controlled not so much by bulk lipophilicity but instead by the fleeting energetic cost of dehydrating ordered water molecules attached to polar functional groups as the solute partitions into the membrane's hydrophobic interior; alkyl linkers avert this cost by providing a smooth hydrocarbonic surface with limited hydrogen bond acceptors for the surrounding solvent. The lack of repeating ether oxygens obviates the high energy water cages that would otherwise envelop PEG segments, collapsing the energetic barrier between the aqueous and lipid phases and accelerating passive diffusion. In addition, the conformational freedom of saturated chains enables the molecule to achieve a compact, low profile shape in the bilayer, minimizing the area penalty required to deform the membrane. Branching may be incorporated without re-introducing polarity: a single methyl group around the mid point induces a slight kink that perturbs crystallinity in the solid state without significantly altering the hydrophobic surface experienced by the membrane. Unsaturation may be incorporated sparingly to confer rigidity necessary for ternary-complex geometry without re-introducing the polar surface area that would result from an analogous insertion of ether units. The combined effect is an increase in trans-cellular flux observable in artificial membrane assays as well as in intact epithelial monolayers, enabling an earlier onset of intracellular target degradation without the need for active uptake transporters whose polymorphic expression can confound inter-species scaling.
Increased Lipophilicity for Bioavailability
Oral bioavailability is often limited by a balance between lipophilicity (favoring membrane partitioning) and metabolic liability (induced by excessive lipophilicity); alkyl linkers walk this line by providing a reliable, linearly-tunable logP that increases with the number of methylene groups and is agnostic to the electronic properties of the pendant ligands. Since hydrocarbon oxidation is predominantly ω-terminal, increased chain length does not greatly accelerate clearance: instead, the resulting single hydroxy metabolite is only slightly more polar and often retains activity, rather than abruptly losing exposure as when PEG chains are truncated at any site. Moreover, the lack of ether bonds also eliminates peroxide-prone weak points that may lead to chain fragmentation in vivo, so the shape of the PK profile is dictated by one dominant, predictable biotransformation rather than by a statistical distribution of truncated homologues. The addition of an alkyl tether to the formulation allows lipid-based drug-delivery vehicles such as self-emulsifying systems, lipid nanoparticles or cyclodextrin complexes to transport the degrader without needing high ionic strength or pH conditions that could cause chemical degradation. Combined, alkyl linkers offer a straightforward, synthetically-accessible strategy to boost tissue exposure while preserving the structural integrity required for reproducible pharmacology.
Reduced Polarity Compared to PEG
Polarity, however, is not simply the sum of heteroatoms: It also communicates information about the directionality of partial charges and their ability to scaffold surrounding water molecules into relatively structured shells. Alkyl linkers blunt this effect by offering a uniform hydrocarbon surface whose dielectric constant approaches that of the molecule itself, thereby smoothing out the electrostatic decay that would otherwise present a kinetic barrier to partitioning. Replacing each ether oxygen with an isosteric methylene unit extinguishes a permanent dipole and two hydrogen-bond acceptors, reducing topological polar surface area in increments that are undetectable by the binding site but highly salient to the lipid bilayer. The spoliation of polar functionality likewise reduces the probability of efflux recognition: Substrates of P-glycoprotein and BCRP often carry a minimum complement of hydrogen-bond acceptors for high-affinity engagement, a standard that alkyl-rich degraders cannot meet, so net flux is determined by passive diffusion rather than by active extrusion. The dampening of polarity also weakens non-specific binding to plasma proteins mediated by charge-dipole interactions, releasing a larger unbound fraction that can more easily equilibrate into tissues. Alkyl linkers serve as low polarity alternatives to hydrophilic PEG scaffolds which give medicinal chemists a way to traverse between polarities while maintaining modular synthesis pathways essential to contemporary drug development.
Case Studies and Literature Evidence
In the degrader campaigns that are publicly available, 3- or 5-methylene hydrocarbon chains have been used multiple times to salvage programs in which PEGylation has hit its polarity limit. The shorter linker reduces topological polar surface area without the molecular-weight cost of a long ethylene-glycol chain and can thus facilitate rapid access to tumour or neuronal tissue, yet can still retain the exit vectors needed for catalytic ubiquitination. In head-to-head comparisons that have been published, the same warhead–E3 pair can swing from cytostatic to cytotoxic as the tether is reduced from PEG6 to C5, and this is attributed to faster intracellular build-up rather than improved binding affinity. In any case, the literature highlights that context is key: alkyl dominance appears when the therapeutic index is limited by permeability or when high-affinity ligands are themselves insoluble, while PEG maintains its throne for targets where an extended ternary-complex half-life is required. The overall message is therefore empirical rather than ideological – the biological choke-point, not dogma, should guide linker choice.
C3, C5 Linkers in Cancer PROTACs
Initial structure–permeability relationships of bromodomain and androgen-receptor degraders first identified the C3 propyl group as the shortest hydrocarbon moiety capable of reclaiming activity in suspension cell lines that had been previously unyielding to their PEGylated counterparts. Superimposition of co-crystal structures demonstrated that this concise alkyl segment maintains the pharmacologically active conformation (i.e. does not introduce deformation) by simply ejecting a single ordered water molecule at the ligase interface, modestly condensing the ternary complex and offsetting the entropic penalty of rotor immobilization upon complex formation. Extending to C5, the pentyl moiety confers one additional rotameric shell. However, a C2 branch via a gem-dimethyl motif shrinks this extended architecture down to an effective length on par with C3 while contributing lipophilicity that further boosts membrane permeation. In castration-resistant prostate cancer models, the C5-appended degrader maintained intratumoural concentrations above the cellular DC50 across an entire dosing cycle, while the equivalent PEG counterpart plummeted below the concentration threshold within hours, a distinction ascribed to diminished active efflux rather than differential metabolic turnover. Crucially, neither linker length triggered colloidal precipitation in cassette-style pharmacokinetic assessments, highlighting that this increase in permeability is not compensated by formulation instability. Taken together, these cancer-centric narratives establish C3 and C5 as the default alkyl yardsticks: long enough to shield polarity, short enough to dodge microsomal snares, and geometrically innocuous to the subtle dance of induced-fit proximity.
When Hydrophobic Linkers Outperform PEG
Systematic comparisons have found hydrophobic alkyl tethers to outperform PEG in cases where the molecular set already contains an excess of hydrogen bond donors, or where the therapeutic window is governed by efflux rather than potency. Under these conditions, the PEG surface decorated with ether oxygen "tags" the degrader for recognition by ABC transporters, and actively transports the degrader back to the outside of the cell before it can engage its target. Substitution of the glycol repeat with a saturated alkyl chain obliterates these telltale acceptor atoms, thus "flattening" transporter affinity, and moves the rate-limiting step from the permeation–efflux equilibrium to simple passive diffusion. A second case where alkyl is preferred to PEG arises when the E3 ligand is itself heavily fluorinated or carbocyclic; in this instance, the addition of a hydrophilic PEG chain increases molecular size without enhancing solubility, yielding waxy oils at ambient temperature that are difficult to filter and lyophilize. The alkyl analogue, being small and non-hygroscopic, readily crystallizes, and meets residual solvent limits without "chasing." Finally, in oral administration strategies, the reduced polar surface conferred by alkyl sidechains is a better match for the "lipid rail" routes used by intestinal lymphatics, and so enables absorption through chylomicron "hitchhiking" that avoids first-pass metabolism in the liver. In a series of independent publications, these and other related factors have come to give hydrophobic linkers the upper hand, especially against targets present in sanctuary tissues where efflux activity is abundant and aqueous humor is scarce.
Considerations in Choosing Alkyl Linkers
Choosing an alkyl tether is less a matter of its lipophilicity directly than of tuning the chain length so that solubility, permeability and ternary-complex geometry all point to the same answer. Since each methylene group increases membrane affinity but decreases aqueous solubility, the choice must take into account early predictions of solubility along with scores for efflux risk and anticipated metabolic clearance, or the first in vivo trial may identify a molecule that traverses membranes rapidly but precipitates from gastric fluids or sequesters irreversibly in fat tissue. For this reason, successful programs consider the alkyl linker a multivariate parameter whose chain length, branching and heteroatom punctuation are varied in conjunction with the polarity of the ligand and E3 components, so that the final construct falls in a physicochemical space compatible with both oral absorption and sustained plasma exposure.
Balancing Solubility and Permeability
The alkyl chain imparts a continuous increase in lipophilicity that can redress the deficit in permeability when the pharmacophores are already over-endowed with H-bond donors, but at the same time the hydrophobic tract entropically disfavors hydration and structurally predisposes towards crystallization in biorelevant media. A practical compromise is achieved by incorporating a single, weakly basic N or a tertiary alcohol at some position in the linker; these groups provide an anchor for protonation or transient H-bonding that boosts kinetic solubility at stomach pH without reanimating the polar surface area that would otherwise be detrimental to membrane permeation. Branching has a two-fold effect: gem-dimethyl substitution at or near the middle of the chain impedes the packing of extended alkyls, thus precluding micellization, and the resultant steric congestion simultaneously constricts the conformational space so that the effective cross-sectional area presented to the lipid core is smaller. Placing the branch next to an sp² centroid (e.g. an aromatic ring or an amide) further limits the conformational space and biases the exit vector to the bound conformation, which compensates for the entropic cost of desolvation. Repeated turbidimetric and artificial-membrane assays show that these subtle changes widen the margin between precipitation threshold and permeability ceiling, which allows the chain to be extended until efflux is outsmarted without overshooting into insolubility. The result is a structure whose dissolution rate in fasted-state simulated fluid is larger than the flux needed for absorption, while the apparent permeability is close to the asymptotic level expected for trans-cellular diffusion, a synergy that typically leads to detectable oral exposure in higher species without resorting to enabling formulations that burden late-stage development.
SAR Optimization Insights
Structure–activity relationships focused on the alkyl linker are also different from those of traditional ligand optimization: the tether itself is not identified by either protein surface, instead being responsible for tuning distance, vector angle, and physicochemical signature. Systematic evaluations have shown that cooperativity is often optimal when the chain length positions the two warheads at an end-to-end distance that matches the average separation observed in cryo-EM ternary complexes, but cellular activity can continue to rise one or two carbons past this crystallographic sweet spot, suggesting that membrane permeation and intracellular free fraction (not just binding thermodynamics) dictate the observed DC50. Branching every third carbon produces a "silky" alkyl surface that is not recognized by cytochrome oxidases, extending half-life without adding polar metabolic handles that would reinstate efflux susceptibility. If the ligand itself is heavily fluorinated, shortening the alkyl linker by one carbon and inserting a single ether oxygen at the midpoint can recover solubility while maintaining the low polar surface area required for passive diffusion; such mixed motifs outperform either pure PEG or pure alkyl analogues, highlighting that the SAR is not linear but sigmoidal with respect to hydrophobe/hydrophile ratio. Finally, because alkyl tethers are metabolically inert toward ether-cleaving enzymes, structure–activity trends observed in vitro will be recapitulated in vivo with fewer surprises, allowing groups to home in on an optimal chain length and branching pattern within two synthetic iterations rather than the multi-cycle campaigns historically required for PEGylated degraders.
Supply & Customization Options
At BOC Sciences, we supply a complete portfolio of alkyl linkers designed to improve the lipophilicity and membrane permeability of PROTAC molecules. Each linker is produced under strict quality control and comes with full analytical documentation to ensure consistent performance across your screening or lead-optimization campaigns.
Chain Length Variants (C3–C12)
We offer a broad selection of alkyl chain variants ranging from C3 to C12, covering the most commonly used linker lengths in PROTAC research.
- Shorter chains (C3–C5) provide greater flexibility and are suitable for early exploration studies or ligase–target pairs that require shorter spatial separation.
- Medium chains (C6–C8) deliver a balanced profile of permeability and solubility, often used in lead optimization.
- Longer chains (C10–C12) enhance lipophilicity for systems where deeper membrane penetration or hydrophobic interactions are advantageous.
All linkers are available in high purity (≥98%), confirmed by HPLC, LC-MS, and NMR, and shipped with a detailed Certificate of Analysis (COA).
Requesting Custom Modifications
If your research requires a specific chain length, functional handle, or hybrid structure, we provide custom alkyl linker synthesis tailored to your project's specifications.
Customization options include:
- Functional end groups such as amine, azide, alkyne, NHS ester, or carboxyl.
- Hybrid designs that combine alkyl and PEG elements for improved balance between flexibility and permeability.
- Isotopic labeling or chirality control for advanced pharmacokinetic studies.
Our in-house chemists will assist you in selecting the ideal linker configuration and provide a feasibility quote within 24 hours. Large-scale production and long-term supply agreements are also available for industrial clients.
High-Purity Alkyl Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity Alkyl linkers.
Order High-Purity Alkyl Linkers for Your PROTAC Projects
Alkyl linkers play a critical role in shaping membrane permeability, metabolic stability, and overall PROTAC bioavailability. By carefully selecting the right chain length and chemical composition, researchers can dramatically improve the pharmacokinetic profile of their degraders. At BOC Sciences, we're committed to supporting your discovery workflow with reliable, high-purity alkyl linkers - from C3 to C12 - and flexible custom synthesis options.
Get in touch with our technical team today to request a quote, discuss your linker design, or explore custom modifications for your next-generation PROTAC program. Let's accelerate your path from concept to clinical candidate with chemistry you can trust.
FAQs
1. Why use alkyl linkers in PROTACs?
Alkyl linkers increase membrane permeability and hydrophobicity, improving compound uptake and cellular bioavailability.
2. What chain lengths are commonly used?
C3, C5, C6, and C8 linkers are typical, providing tunable hydrophobicity and linker flexibility.
3. Do alkyl linkers reduce solubility?
They may, but this can be balanced with hybrid PEG–alkyl structures for optimal physicochemical profiles.
4. Do you supply alkyl linkers?
Yes, we offer C3–C12 linkers in high purity, available for bulk and custom synthesis requests.
References
- Zografou-Barredo N A, Hallatt A J, Goujon-Ricci J, et al. A beginner's guide to current synthetic linker strategies towards VHL-recruiting PROTACs[J]. Bioorganic & Medicinal Chemistry, 2023, 88: 117334. https://doi.org/10.1016/j.bmc.2023.117334.
- Troup R I, Fallan C, Baud M G J. Current strategies for the design of PROTAC linkers: a critical review[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 273. https://doi.org/10.37349/etat.2020.00018.
- Kelm J M, Pandey D S, Malin E, et al. PROTAC'ing oncoproteins: targeted protein degradation for cancer therapy[J]. Molecular Cancer, 2023, 22(1): 62. https://doi.org/10.1186/s12943-022-01707-5.
- Liu Z, Zhang Y, Xiang Y, et al. Small-molecule PROTACs for cancer immunotherapy[J]. Molecules, 2022, 27(17): 5439. https://doi.org/10.3390/molecules27175439.
- Distributed under Open Access license CC BY 4.0, without modification.
