Choosing between a three-, five- or eight-carbon alkyl tether is not an all-or-nothing proposition based on the three structures published to date. Rather, the range of linker lengths is an adjustable hydrophobic fulcrum. C3 is the equivalent of a short-length molecular ruler, which pulls the POI-E3 pair into a low-nM, fast-on/fast-off complex. This is ideal for cytosolic targets with narrow egress channels. C5 is the middle option, which adds one more set of rotamers to accommodate subtle discrepancies in binding-site spacing without occluding the breathing motion necessary for processive ubiquitination. C8, in contrast, trades enthalpic grasp for hydrodynamic size, a tradeoff that can remedy plasma half-life or broaden hook-effect-limited capacity, but can also ablate effective molarity below the level needed for catalysis. The practical rule of thumb learned so far from both cancer and neurodegenerative disease programs is thus one of trial and error, rather than edict: start with the shortest alkyl that doesn't cause aggregation, progress to the next homologue if biochemical efficacy plateaus, and only go back down if lipophilicity-associated toxicity or solubility issues force it.
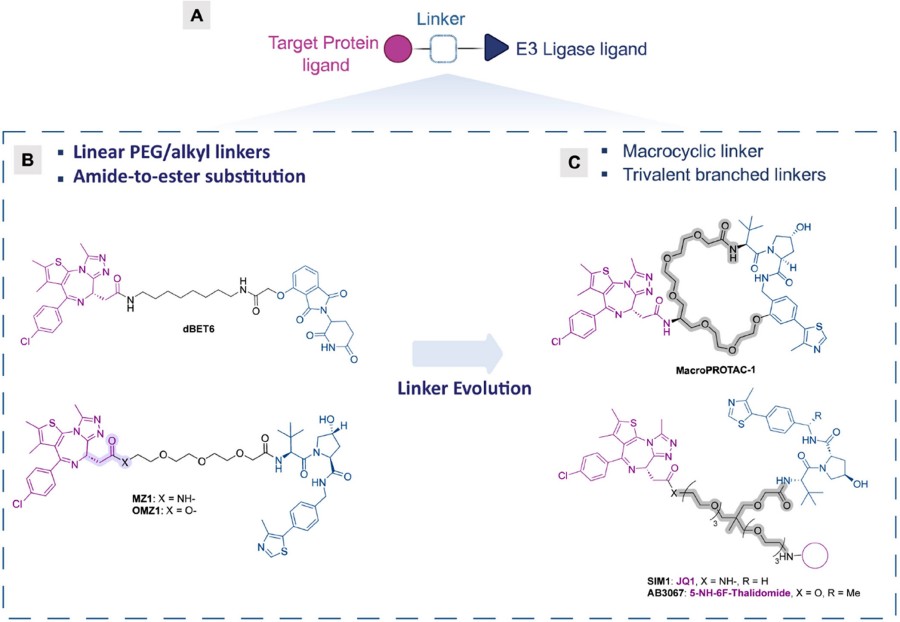 Fig. 1 Representative examples of PROTAC linkers.1,2
Fig. 1 Representative examples of PROTAC linkers.1,2
Why Alkyl Chain Length Matters?
The methylene unit count in the linker affects how the PROTAC compromises three competing equilibria: its partition coefficient into lipid bilayers, the ordering of water molecules around the ligand–ligase interface, and its susceptibility to oxidative truncation. Too-short a chain immobilizes the ternary complex too stringently, entropically penalizing the sampling of lysines; too-long a chain reduces the effective molarity of the two reacting moieties, and also predisposes the complex to intramolecular hydrophobic collapse. Since each methylene provides a lipophilicity increment that is close to a constant value, linker length can be optimized in small steps, rather than in the quantum jumps required by the discrete PEG homologues. The same incremental reasoning applies to metabolic stability: while unbranched alkyl chains of longer length are more likely to be β-oxidized, odd-numbered tethers (C3, C5) are partially shielded from the classical spiral, since the final thiolysis product is a propionyl-CoA that is shuttled into anaplerotic rather than catabolic pathways. In this way, linker length is not just a passive spacer property; it is a vector that can be tuned to connect membrane transit and intracellular lifespan.
Impact on Permeability
Short alkyl tethers (C3) minimizes the hydrophobic surface area and reduces the time taken to diffuse through the lipid bilayer. It also avoids the micellar entrapment that is more likely to be an issue for long hydrocarbon chains. The methylene spine only samples σ-bond rotation, so the entity is more like a semi-flexible rod than the entropic spring provided by PEG; this can be a positive if the POI and E3 surfaces are a fixed distance apart, because the entropic cost of freezing the spacer into one productive conformation is less. C5 adds one more rotamer set that can tolerate some diversity in gate-keeper residue identity, which is lost when the chain is reduced to C3. C8 gives more reach, but the two extra carbons also start to reintroduce the hook effect: the molecule can stabilize POI–PROTAC or E3–PROTAC binaries instead of the productive trimer, and the increased hydrodynamic radius subjects it to faster renal clearance than cellular internalization. The experimental result is thus an observed permeability optimum that often occurs at C5 when the lipophilicity is matched, a behavior that is consistent in both cancer and neurodegeneration cell lines.
Effects on Solubility and Stability
Alkyl linkers lack the ether oxygens that order hydration shells, so solubility must be designed in another way – typically salt formation, amorphous dispersion or co-solvent systems. C3 is unlikely to compete with these approaches because the hydrophobic increment is small; C5 begins to rival polar ligand motifs for solvation, and C8 can push the whole package into a colloidal precipitation region unless a basic center or morpholine tail is added to restore hydrogen-bond donation. Metabolic stability provides a flip side to this story: probability of β-oxidation rises with unbranched chain length, but odd-numbered tethers (C3, C5) escape the classical spiral to a degree because the last thiolysis leaves a propionyl-CoA residue that is shunted into anaplerotic rather than degradative pathways. C8 by contrast will undergo two rounds of β-oxidation to yield acetyl-CoA, truncating plasma half-life unless a gem-dimethyl block is added to prevent cytochrome binding. Taken together, the solubility–stability scorecard identifies C5 as the compromise median: long enough to increase membrane affinity, but short enough to be compatible with traditional formulation carriers and to escape oxidative truncation that would otherwise require heroic formulation interventions.
Comparative Review
The activity profiles of C3, C5 and C8 hydrocarbon spacers of otherwise identical degraders reveal a parabolic response with the apex at C5. The shortest chain provides the most flexible conformational response and the most rapid permeability onset but it often suffers from insufficient reach to promote high-affinity ternary complex formation, whereas the longest chain provides the deepest lipophilicity and the longest residence, at the cost of a significant drop in solubility, increased microsomal clearance and increased efflux recognition. Case reports in medicinal chemistry therefore tend to focus on the pentyl tether as the pragmatic compromise while C3 or C8 analogues are used as strategic counter-scaffolds to stress-test the robustness of structure–activity hypotheses.
C3 Linkers: Short, Favorable for Flexibility
A three-carbon alkyl bridge is the closest thing to a molecular hinge. Its low steric demand allows very fast rotation about all σ-bonds, which means that the warheads can access a broad conical volume and sample a range of encounter geometries with the target and ligase surfaces. This flexibility is favorable in cases where available crystal structures indicate that the two proteins need to "close" a significant distance upon complex formation; the short linker reduces the entropic cost of fixing an extended chain, often resulting in faster association kinetics and observable degradation at lower cytosolic concentrations. Synthetic-wise, C3 can be introduced by standard alkylation or cross-metathesis without the need for protecting-group acrobatics, and its low molecular weight has negligible impact on the overall mass of the conjugate. The obvious limitation is that the same brevity might preclude the degrader from spanning the physiologically relevant separation, resulting in partial ternary complexes with marginal cooperativity and fast off-rates. On the other hand, the modest increase in lipophilicity may be insufficient to rescue compounds already carrying polar heteroatoms, so cellular uptake is modest in the absence of active transporters. However, early hit-to-lead campaigns often retain a C3 option as a "flexibility benchmark" against which longer, more rigid analogues are compared, so that any gain in potency observed downstream can be ascribed to improved reach rather than to fortuitous binding entropy.
C5 Linkers: Balanced Properties
Pentyl chains straddle the sweet spot between permeability, solubility and synthetic tractability. The two additional methylenes over C3 double the lipophilic increment, nudging the molecule over the hysteresis threshold for effective passive diffusion, while the chain remains short enough to preclude the self-association and crystallinity that complicate longer hydrocarbons. Conformationally, C5 straddles the line between extended and mildly kinked geometries; this flexibility enables the degrader to fit both "open" or "closed" ternary-complex configurations without inducing a disfavored torsion on the warheads. Critically, ω-oxidation remains the principal metabolic pathway, but the resulting alcohol is generated at a rate that seldom outpaces hepatic blood flow, so clearance is instead perfusion-limited. In formulation studies, C5-linked compounds dissolve readily in simulated gastric fluid without the need for pH modifiers or cyclodextrin complexes, and they remain miscible in lipid-based self-emulsifying systems, making it a versatile platform for both oral and parenteral formulations. All together, these properties help explain why C5 is so ubiquitous in the patent literature as the "first-choice" alkyl spacer: it extends the reach of the degrader, amplifies membrane affinity and still exhibits small-molecule-like behavior during salt selection and polymorph screening.
C8 Linkers: Enhanced Lipophilicity, But Risks in Solubility
Octyl tethers provide the greatest apparent permeability of the homologous series, at the cost of an exaggerated loss of aqueous solubility and greater non-specific tissue sequestration. The longer eight-carbon span promotes the formation of a hair-pin conformation in polar media, sequestering the hydrocarbon core and orienting the polar warheads toward the solvent; this conformation is entropically favored in water, but it increases the effective molecular footprint and the phase separation rate. Crystallinity data show that C8 conjugates preferentially crystallize in high melting polymorphs that are difficult to wet, so dissolution is rate-limiting even at small particle size. From a metabolic perspective, the longer chain provides two terminal methylenes that are substrates for ω-oxidation, which doubles the clearance probability and creates a diacid metabolite whose polarity is sufficient to promote renal elimination but whose activity is often reduced. Biologically, the increased lipophilicity also promotes partitioning into fatty and myelin-rich tissues, resulting in a large volume of distribution and a long terminal phase that muddles dosing. The C8 linker is still often used as a pharmacological probe: when the ternary complex requires an extended reach, or when the target is located in a membrane micro-domain, the octyl tether may provide the necessary span and residence time. Teams will usually synthesize the C8 analogue once the C5 lead is identified, and then use it to stress test the upper bounds of the structure–activity envelope, to confirm that any shortening or branching strategy will land safely within developable space.
Literature Examples and Experimental Evidence
A compendium of recent peer-reviewed publications in which only alkyl chain length was intentionally varied and thus is the only structural determinant that may impart unique influence on potency degradation, permeability and PK behavior provides side-by-side comparison data. For many target classes (kinases, epigenetic readers, nuclear receptors, etc. ), C5 is often the most common and frequently-validated "balanced" tether while C3 and C8 analogues are often concurrently synthesized as a boundary conditions to stress test the robustness of any resultant structure–activity relationship. The growing body of work is illustrative that each incremental addition of methylene groups does more than increase reach, it also re-writes the desolvation landscape, modifies efflux liability and re-maps the metabolic landscape converting the linker from a passive spacer into an active driver of developability.
NanoBRET-Based Intracellular Engagement Screen
A cellular high-throughput assay, which pairs bioluminescence resonance energy transfer (BRET) to Nano-luciferaze tagging, has been used to measure target occupancy as a function of time, for C3, C5 and C8 alkyl-linked degraders of a single oncogenic kinase. The NanoBRET readout showed that the C3 analogue was rapidly taken up and measurably occupied within minutes of exposure but reached a plateau at a moderate signal level, indicative of a dynamic but suboptimal ternary complex. The C5 congener gave the highest sustained signal, in line with previous biochemical DC50 observations and the C8 compound displayed a initially high read-out which dissipated at a faster rate than expected from simple metabolic turnover; subsequent inhibitor of efflux studies pointed to P-glycoprotein as a cause of the rapid wash-out. Critically, this assay was performed on intact cells without any membrane disruption, so this rank ordering is a convolution of permeability, target occupancy and intracellular stability as opposed to a pure measure of binding affinity. By normalizing the BRET ratio to a cell-impermeable tracer, the authors argued that the superior performance of C5 must be attributable to a more favorable membrane transit/intracellular free fraction profile, an observation that would have been masked if this work were to be performed in a lysate-based quantification protocol only.
Parallel Artificial Membrane and Microsomal Stability Mapping
In a companion study, PAMPA and liver microsomal incubations were carried out on a matrix of C3–C8 alkyl-linked PROTACs using the same warhead. As with PAMPA data, there was an inverted-U pattern: apparent permeability increased from C3 to C5, plateaued at C6 and decreased for C7–C8 as the effective hydrodynamic radius increased. By contrast, microsomal half-life decreased steadily beyond C5 in line with ω-oxidation on both ends when the chain is sufficiently long to present two accessible methyl groups. The authors also observed that C8 generated a diacid metabolite that is almost as polar as the parent PEG-linked degrader, suggesting that too much hydrocarbon extension can bring back the hydrophilic liability that the alkyl strategy was supposed to avoid. Cryo-EM of ternary complexes formed with C5 and C8 linkers showed that the longer chain pushes the E3 ligase into a slightly different orientation, consistent with the biochemical observation that there is no improvement in cooperativity beyond the pentyl length. Taken together, these orthogonal read-outs further support C5 as the inflection point where permeability gains are maximised and solubility, metabolic stability and target engagement are still within developable bounds.
Practical Guidance for Medicinal Chemists
The practical reality in day-to-day decision-making in drug-discovery laboratories is less often influenced by elegant design principles. Instead, there is an overarching hierarchy of rapid, information-rich experiments that triage molecules before the precious resource of in-vivo assets is committed. Skilled teams integrate "early warning" assays for solubility, permeability, microsomal turnover, efflux risk, etc. into the same iterative optimization loop that refines binding affinity so that structure–activity relationships (SAR) are simultaneously structure–property relationships (SPR). The key is to view each new scaffold as a dynamic set of variables: lipophilicity, polar surface, rotatable bonds and ionization state are adjusted in concert, not sequentially, using design matrices that can be interrogated by parallel synthesis and high-resolution mass-triggered purification. By front-loading developability filters and validating them with calibrated in-silico models, chemists avoid the costly cycle of "potency first, liabilities later" and instead converge on candidates whose physicochemical signature is already compatible with oral exposure and scalable synthesis.
Streamlining Multi-Parameter Optimization
A pragmatic workflow starts with a virtual design space that associates 3-D docking scores with real-time ADMET predictions. Hits are ranked not only by affinity but by a weighted composite that takes into account predicted solubility and efflux liability. Parallel one-pot reactions install systematic variations (chain length, heteroatom insertion, branching) and orthogonal protecting groups allow late-stage exchange of warheads without having to re-synthesize the linker. Crude reaction mixtures are analyzed directly by high-throughput turbidimetry and PAMPA so that only fractions that clear both the solubility and permeability hurdles are purified. This "assay-first" philosophy compresses three traditional cycles into a single laboratory week and also provides an empirical envelope within which more nuanced SAR can be explored with confidence. Importantly, the same analytical files (LC-MS/MS, NMR, ion-mobility) that are used to confirm identity are automatically uploaded to a shared data lake where machine-learning algorithms refine the next round of predictions and gradually diminish the chemical space that must be synthesized to achieve the target product profiles.
Proactive Risk-Mitigation Tactics
Instead of encountering such a late-stage surprise, ideally, an alert team would have coded the alerts for toxicophores and metabolic soft spots into the initial retrosynthetic analyses. Aromatic amines, anilides and Michael acceptors, for instance, can either be eliminated or camouflaged with metabolically unstable protecting groups that only unveil the toxicophore after first pass metabolism. If a phenol is a desired pharmacophore, ortho-methyl or fluoro groups can be introduced to prevent metabolism to quinone-imine intermediates, a strategy that has been demonstrated to remove genotoxicity signals in the Ames test while maintaining potency. Secondary amides that are susceptible to acyl-glucuronidation can also be N-methylated or included as a bicyclic lactam, both maneuvers that stabilize the carbonyl and prevent conjugation while also suppressing idiosyncratic hepatotoxicity signals. Associating these structure-based modifications with early phase high-content screens that are predictive of cytotoxicity (mitochondrial membrane potential, bile-acid export, steatosis imaging, etc.) gives a detective readout of potential liabilities while the molecular weight is still low enough to enable wholesale reengineering if necessary. The result is a group of candidates whose margins of safety can be interrogated well before time- and resource-intensive two-year animal studies, thus shortening timelines and reducing the attrition that has historically challenged first-in-human decision points.
Ordering Options for C3-C8 Linkers
At BOC Sciences, we provide a comprehensive range of C3-C8 alkyl linkers specifically optimized for PROTAC synthesis and structure-activity relationship (SAR) exploration. Each linker is validated for high purity, and supplied with full analytical documentation to ensure consistent performance in every experiment. We maintain ready-to-ship inventory for the most commonly used variants - C3, C5, and C8 linkers - making it easy for research teams to quickly access critical materials without long lead times. Every batch is backed by a Certificate of Analysis (COA) confirming identity, purity, and analytical results (HPLC, LC-MS, NMR).
Flexible Supply to Match Your Research Needs
- Small-scale quantities (100 mg - 1 g): Ideal for screening, linker optimization, or proof-of-concept studies.
- Medium- to large-scale quantities (10 g - multi-kg): Suitable for advanced development and preclinical manufacturing.
- High-purity custom batches: Tailored for GMP-adjacent or regulatory-grade applications.
Custom Synthesis and Modification Services
Need a unique variant for your program? Our custom synthesis service allows you to:
- Adjust chain length (C2-C12 or intermediate variants).
- Modify functional groups (amine, azide, alkyne, NHS, carboxyl, or hybrid PEG-alkyl structures).
- Request analytical support, stability studies, or isotopic labeling for pharmacokinetic analysis.
We also offer technical consultations with our chemists to help you select the most suitable linker length and structure based on your target protein, E3 ligase, and permeability requirements.
Fast Global Delivery
All linkers are packaged with stability in mind and shipped worldwide through trusted logistics partners. We support temperature-controlled shipping and provide full tracking for every order, ensuring safe and timely delivery to your lab. Contact us today to request a quote, check stock availability, or discuss a custom synthesis project for your next PROTAC study. Our team will provide a tailored solution within 24 hours, helping you accelerate discovery with chemistry you can rely on.
High-Purity Alkyl Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity Alkyl linkers.
FAQs
1. Which alkyl linker length is most effective?
C5 linkers often strike the best balance between flexibility, permeability, and solubility, but the optimal choice depends on the specific protein-ligase system.
2. How does chain length affect PROTAC properties?
Shorter linkers offer rigidity, while longer ones enhance lipophilicity and membrane penetration.
3. Can I order C3-C8 linkers in bulk?
Yes, we provide in-stock C3, C5, and C8 linkers with flexible MOQs and custom scale-up options.
4. Do you offer analytical documentation?
Every batch includes a COA, HPLC, LC-MS, and NMR data for full traceability.
References
- Image retrieved from Figure 1 " Representative examples of PROTAC linkers," Salerno A.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Salerno A, Wieske L H E, Diehl C J, et al. Rational Design of PROTAC Linkers Featuring Ferrocene as a Molecular Hinge to Enable Dynamic Conformational Changes[J]. Journal of the American Chemical Society, 2025, 147(16): 13328-13344. https://doi.org/10.1021/jacs.4c18354.
- Troup R I, Fallan C, Baud M G J. Current strategies for the design of PROTAC linkers: a critical review[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 273. https://doi.org/10.37349/etat.2020.00018.
- Anwar Z, Ali M S, Galvano A, et al. PROTACs: the future of leukemia therapeutics[J]. Frontiers in Cell and Developmental Biology, 2022, 10: 851087. https://doi.org/10.3389/fcell.2022.851087.
- Burke M R, Smith A R, Zheng G. Overcoming cancer drug resistance utilizing PROTAC technology[J]. Frontiers in Cell and Developmental Biology, 2022, 10: 872729. https://doi.org/10.3389/fcell.2022.872729.
