Nonpolar alkyl linkers are making a comeback in the world of medicinal chemistry as hydrocarbon-rich, modular tethers that sacrifice aqueous solubility for accelerated membrane permeability. In addition to removing recurring ether oxygens to reduce the topological polar surface area, they help to sharpen the lipophilicity gradient and push the entire PROTAC to pack into a smaller, more cylindrical form in the lipid bilayer. The same features also simplify large-scale manufacture—malonate extension or cross metathesis is enough—and reduce oxidative liabilities, since methylene backbones are unreactive toward peroxidases or cytochrome P450 oxygen rebound. As a result, alkyl spacers are no longer dismissed as vestigial linkers from first-generation tool compounds; they are now judiciously re-employed during lead optimization once the ternary-complex geometry is set and the main optimization hurdle is systemic exposure rather than target engagement.
Introduction to Hydrophobic Alkyl Linkers
Hydrophobic alkyl linkers are linear or branched hydrocarbon chains that tether a target-binding warhead and an E3-recruiting ligand without the recurring ether oxygens. The lack of polar heteroatoms reduces the dielectric constant along the spacer and this can bias the whole construct towards a more compact and membrane compatible conformation upon exiting the aqueous phase. These linkers are also synthetically attractive since they can be built from commodity di-acids, amino-alcohols or terminal alkenes by routine amide, ester or cross metathesis, thus avoiding the chromatographic desalting steps required for PEG homologues. The main liability of alkyl spacers—reduced aqueous solubility—is usually remedied by salt formation or by capitalizing on the intrinsic polarity of the adjacent ligands, a trade-off that is acceptable in a situation where the therapeutic goal is rapid intracellular accumulation, rather than prolonged systemic retention.
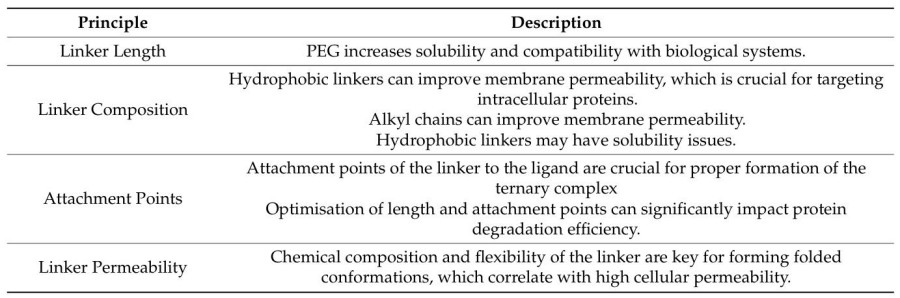 Fig. 1 Principles of linker design in PROTACs.1,5
Fig. 1 Principles of linker design in PROTACs.1,5
Why Hydrophobic Linkers Are Important in PROTACs?
The underlying dogma here is that the catalytic ubiquitination reaction only occurs when the POI and E3 ligase are constrained in a transient, favourably-oriented encounter. Hydrophobic linkers speed up the first half of this choreography—the collision frequency—by shuttling the PROTAC across the lipid bilayers at a rate hydrogen-bond-rich spacers can't compete with. In the cytosol, the same hydrocarbon surface repels structured water from coating the ternary complex, effectively reducing the entropic penalty of immobilizing the linker into a productive orientation. This double life—membrane taxi and conformational lubricant—explains why many programmes see a left-shift in the cellular DC50 when the ether-rich tether is replaced by a 4–6 carbon alkyl, even when the in vitro binding affinity is unaltered. It also opens up the metabolic vulnerabilities of alkyl chains (terminal ω-oxidation), which are well mapped out, allowing teams to introduce gem-dimethyl branches or cis-double bonds that prevent the undesired cleavage without restoring polarity. Precedents from lipid-based pro-drugs and oral small molecules further de-risks the safety discussion, because reviewers already accept that saturated hydrocarbon fragments are unlikely to produce reactive metabolites. As a result, hydrophobic linkers have gone from being specialist tools for brain-penetrant agents into mainstream options for any target where uptake kinetics, rather than ternary-complex residence time, is the rate-limiting constraint.
Key Differences from PEG-Based Linkers
PEGs are water-soluble due to the repeating ether oxygen's lone pairs, which are able to order surrounding water into hydrogen bond networks. Hydrocarbon linkers, by contrast, are "lipophilic" by default because they eschew that offering to bulk water. This is not mere semantics, as these two strategies have distinctly different profiles across 4 parameters.
- (1)Permeability: PEG linkers increase topological polar surface area and often result in active efflux, while hydrocarbon linkers mute both and are conducive to passive diffusion.
- (2)Metabolism: PEG linkers can be cleaved by peroxidation, which can generate a distribution of metabolites that complicate PK interpretation. Oxidation of alkyl linkers, by contrast, is often predictable and occurs at terminal methylenes, so that they generate a single alcohol or acid metabolite, which is often equipotent.
- (3)Solubility: PEG linkers are susceptible to disruption of their entropy-driven hydration by high ionic strength or co-solvent perturbation, while hydrocarbon linkers are soluble only with formulation assistance (cyclodextrins, lipid-based) and are less affected by ionic milieu.
- (4)Analytical ease of characterization: polydisperse PEG molecules contribute to peak broadening and spectral confusion by mass spec, while monodisperse hydrocarbons contribute sharp, isotopically resolved peaks and simplify the ability to set specifications. In addition, from a synthetic perspective, hydrocarbon chains are often straightforward to elongate using commodity reagents and familiar coupling chemistries, while high-molecular-weight PEGs often require anhydrous conditions and metal catalysts.
As such, the choice between PEG and hydrocarbon is not simply a matter of adjusting the logP dial, but can have a profound impact on manufacturability, metabolic robustness, regulatory specifications, and in turn, clinical exposure.
Core Advantages of Hydrophobic Linkers
Hydrophobic alkyl linkers provide a physicochemical tool kit that directly tunes the most frequent attrition factors in bifunctional degrader campaigns: poor membrane permeability, sub-optimal oral exposure, and rapid renal clearance. By discarding the repeating ether oxygens that bloat PEG chains with structured water, hydrocarbon spacers decrease topological polar surface area in predictable methylene increments, steepen the correlation between passive permeability and unbound fraction, and collapse the construct into a cylinder that can navigate through tight epithelia without recruiting efflux transporters. The same hydrophobic exterior also mitigates intramolecular hydrogen bonding, freeing the two warheads to "search" protein surfaces without becoming kinetically trapped in self-solvated conformations. Consequently, teams that implement C4–C6 alkyl tethers often experience a left-shift in cellular DC50 and a concomitant extension of oral half-life, benefits that can outweigh the solubility penalty once salt formation or nanoparticle dispersion is integrated into the formulation strategy.
Driving Membrane Permeability
Membrane permeability is a function of the transient energetic cost associated with displacing ordered water from polar functional groups. Alkyl spacers address this cost by offering a continuous, low-dielectric surface that the bilayer can "recognize" as "self-like". The lack of periodic ether oxygens abolishes the high-energy water cages that surround PEG chains. As a result, the energetic discontinuity between aqueous and lipid phases is bridged and passive diffusion is enhanced. From a conformational perspective, short alkyl chains inter-sample extended and kinked geometries, enabling the degrader to adopt a compact cross-section upon entering the bilayer while maintaining the reach necessary for an induced-fit binding mode. Branching at C2 or C3 positions further hinders solid-state packing without reviving polarity, which simultaneously improves membrane partitioning and prevents crystallization in gastric fluids. The end result is a quantitative improvement in trans-cellular flux, both in artificial membrane assays and in intact epithelial monolayers. This manifests as a more rapid intracellular target degradation in vivo, without the need for recruitment of active uptake transporters whose polymorphic expression is a frequent source of inter-species scaling issues.
Increasing Bioavailability in Drug Discovery
Oral availability is often limited by a tradeoff between lipophilicity (necessary for membrane partitioning) and metabolic liability (promoted by high grease factors), a space that alkyl linkers bridge by providing a reliable, linearly-tunable logP (increasing by a known increment per methylene group) that is agnostic to the electronics of the tethered ligands. Since hydrocarbon oxidation is ω-terminal, elongating the alkyl chain length doesn't result in dramatic increases in clearance as the metabolite will simply be the one hydroxy metabolite with a relatively small increase in polarity and often with retained activity, whereas cutting PEG chains at random positions can lead to a sudden loss in exposure. Additionally, the high lipophilicity provided by the alkyl tether allows the degrader to be loaded into lipid-based drug-delivery systems such as self-emulsifying matrices, lipid nanoparticles, or cyclodextrin complexes without resorting to extreme pH or high salt concentrations that may promote chemical instability. For these reasons, alkyl linkers provide a simple, synthetically-accessible approach to increasing tissue exposure.
Balancing Lipophilicity and Solubility
The primary objective of alkyl design is to capture the benefit of increased membrane permeability without sacrificing solubility in stomach contents or becoming stored in fat. Addressed by many teams, one approach is to include a single, weakly basic nitrogen or a tertiary alcohol on the alkyl chain; these "handles" can be protonated or engage in transient hydrogen bonding to boost kinetic solubility in the stomach without re-introducing polar surface area that would impede passage through the membrane. Branching can also be used, since gem-dimethyl substitution in the middle of the chain reduces extended alkyl packing and thus formation of micelles, and the added steric bulk also reduces the cone angle, such that the overall cross-section to the membrane's lipid phase is decreased. Iterative testing with turbidimetry and artificial-membrane techniques will show that these small changes widen the range between solubility and permeability limits such that it is possible to extend the chain length until efflux is no longer an issue without hitting the ceiling of solubility. The result is a molecule whose solubility (rate of dissolution) in fasted state simulated intestinal fluid is above the threshold for absorption, and whose apparent permeability is near the saturation value expected for passive diffusion, yielding a compound that can be developed without formulation to result in oral systemic exposure in advanced species.
Customization Options for Researchers
In stark contrast, hydrocarbon linkers can be mixed-and-matched with Lego-like modularity that ethylene-glycol backbones can't match: each methylene can be added or subtracted without changing the electronics of neighboring amide bonds, and the same synthetic step that installs C3 can be extended to C12 simply by exchanging feed-stock equivalents. Because the growing chain is hydrophobic, protecting-group strategies are similarly modest—Boc or Fmoc handles can survive malonate elongation and the terminal carboxyl or amino remains available for late-stage diversification. The resulting palette (C3–C12) maps almost linearly to lipophilicity, letting teams sculpt permeability in calibrated strokes rather than the quantum jumps imposed by discrete PEG oligomers. End-group identity is equally plastic: the same alkyl bromide that seeds chain growth can be displaced in one pot by azide, thiol, or maleimide nucleophiles yielding, orthogonally reactive handles that click directly onto ligand cores without additional amidation steps. Finally, the entire hydrocarbon segment can be "salt-and-peppered" with single ether, alkene or cyclopropyl units to create hybrid hydrophobic–polar motifs that retain the permeability dividend while disrupting crystalline packing in the solid dosage form. In short, researchers can gain access to a combinatorial playground where length, branching, heteroatom placement and terminal functionality can be mixed-and-matched under standard glassware, producing bespoke linkers whose analytical signatures remain monodisperse and whose regulatory precedents are already accepted.
Chain Length Variability (C3–C12)
Chain length is a three-dial tuning knob: each methylene increment pushes both reach and logP deeper and constricts the rotational envelope. C3 provides the most agile conformation, which can span the shortest target-dividing cleft but has shallow enough lipophilicity increase to remain amorphous in initial DMSO solutions. Progression to C5–C6 positions the drug near the top of the permeability bell-curve where microsomal oxidation is still ω-terminal and primarily unique, so metabolic loss is forecastable and saltability is not yet challenged. C8–C10 reaches a plateau in membrane flux and starts to engage adipose partitioning and quantifiable efflux detection; these longer lengths are therefore employed in campaigns where the ternary complex necessitates a longer reach or where the payload itself is so heavily fluorinated that a lipophilic ballast is required. C12 approaches surfactant territory, but if applied with only one mid-chain branch or in the presence of a weakly basic heteroatom, it can be diverted from micelle generation and biased toward lymphatic absorption. Crucially, all of the homologous series are accessible from the same ω-functionalized bromoalkane by iterative Wittig or cross-metathesis homologation, so any laboratory can seed a length–activity plot within two synthetic generations and without having to commit to orthogonal protecting-group schemes.
Functional End-Groups for Coupling
The choice of terminal handle is the "passport" that defines which conjugation chemistries can be performed downstream: amide, click, thiol–maleimide, oxime, Suzuki etc. Amine-terminated linkers can be coupled directly to activated esters under aqueous conditions, but that same nucleophile can also be temporarily masked as a phthalimide to withstand brutal organometallic steps, and then deprotected with hydrazine without reacting with the internal ester. Azide caps are useful for bio-orthogonal ligation of cyclooctynes, but also as a stable "stand-in" during palladium-coupling reactions that would reduce a free amine. Carboxylic acids might sound trivial but enable iterative chain extension through Arndt–Eistert homologation, turning a C6 linker into a C7 or C8 building block in one pot. Maleimide and bromoacetamide termini direct cysteine payloads, but that reactivity can be tuned down by an adjacent electron-withdrawing sulfonyl group, giving slow, site-selective conjugation in a "sticky" protein milieu. Carbonate or disulfide end-caps can be chosen for traceless release: those fragments are stable under storage conditions, but fragment under millimolar thiol concentrations to deliver a payload-free metabolite, which can ease regulatory specification. If this choice is made early, and orthogonal tags validated in the same LC-MS sequence that confirms chain length, one can avoid historically costly protecting-group shuffles that can bring linker optimization to a halt.
Hybrid Alkyl–PEG Linker Designs
Chimeras, in contrast, link a shorter hydrocarbon fragment (C4–C6) to a well-defined ethylene oxide "tail" (EO2–EO4). The resulting hybrids self-segregate in lipid vehicles while staying in solution in dilute aqueous preparations. The alkyl half steers membrane permeation and evades Pgp detection, while the PEG cap yields a hydration "cloak" that averts colloidal flocculation in concentrated formulations. Synthetic approaches are convergent: while the alkyl moiety is prepared via textbook Grignard or Wittig reactions, followed by mesylate capping, it is converted to the final product by grafting a single, monodisperse oligo-ethylene glycol onto it via a mild nucleophilic substitution. The linkage is a robust ether, so the chimera is stable to acid/base deprotection steps and the two warheads can thus be independently orthogonalized if necessary. Critically, the hydrophobe/hydrophile transition point can also be positioned on either side of either ligand. This "asymmetric" hydration can thus be used to tweak the microenvironment near the E3 or target-binding interface without a corresponding change to global lipophilicity. Early PK findings in fact show that these chimeras effectively maintain the permeation efficiency of naked alkyls, while displaying the solubility of mid-range PEGs and, in effect, "flattening" the conventional permeability–solubility seesaw. As such, these chimeras are used as late-stage salvage entities when the fully hydrocarbon counterpart is unstable in stomach acid but the fully PEG one is substrapped to efflux.
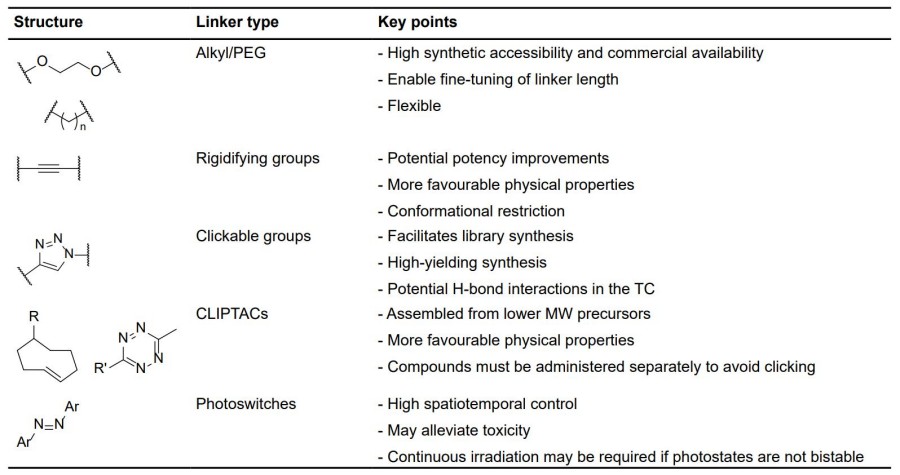 Fig. 2 Key considerations for linker motifs described. 2,5
Fig. 2 Key considerations for linker motifs described. 2,5
Application Areas in Drug Development
Hydrophobic alkyl linker is now commonly seen across the modern R&D landscape. It's one of those chemical insights that's not particularly novel, but which unlocked a tuning knob by which high-affinity ligand pairs could be turned into druggable degraders. The benefit has proven to be most acute in two classes of therapeutic targets, oncology and CNS disorders, in which the limiting pharmacologic barrier is often uptake as opposed to binding. For cancer targets, this "uptake knob" essentially makes it possible for the linker to tune the overall polar surface area of the construct and thus steepen the exposure curve in tumors. For CNS targets, reducing the polar surface area of the molecule likewise speeds the passage across the endothelial layer, without incurring an efflux penalty. On top of that, the linker chain length can be tuned one carbon at a time under standard glassware. As a result, medicinal chemists are able to more rapidly and continuously optimize their candidates, and this optimization in itself conveys both a shorter discovery cycle and a cleaner regulatory narrative. Below we show how the same principle is applied in the areas of mitotic cancer and neurodegenerative proteinopathies, two areas of high unmet need for a rapid and tissue-penetrant knock-down modality.
Oncology and Targeted Therapy
Actionable kinases from solid tumors often have their catalytic clefts located within a helical turn of an E3 surface. A C4–C6 alkyl tether pre-organizes the resulting dyad into a catalytically primed conformation and endows the construct with the lipophilicity needed to partition into the tumor interstitium through enhanced permeability and retention. In the hypoxic core, the hydrocarbon collar prevents ordered water from hydrating the ternary complex, thereby reducing the entropic penalty of lysine presentation and speeding poly-ubiquitin chain elongation. The same linker also protects the molecule from renal peptidases, extending the systemic half-life without requiring a complex nano-formulation that would be difficult to scale up. Recent reports in the literature indicate that a direct exchange of a six-unit PEG for a six-carbon alkyl can transform cytostatic inhibitors into cytotoxic degraders in multiple xenograft models, an effect that was ultimately attributed to more rapid intracellular accumulation rather than better intrinsic affinity. Moreover, the metabolic vulnerabilities of even-numbered chains can be mitigated with the addition of a single gem-dimethyl branch, a modification that prevents β-oxidation without restoring polarity. Taken together, these features make alkyl linkers the de-facto optimization scaffold once the ternary crystal structure indicates that the exit vectors are less than a helical turn apart, a feature that is common to many oncogenic kinases.
Central Nervous System (CNS) Research
Neurodegenerative aggregates are located on the other side of a selectively restrictive endothelium that repels hydrogen bond-rich solutes. Here a C5–C7 alkyl linker reduces topological polar surface area (TPSA) below an empirically defined limit set by P-glycoprotein and allows the degrader to cross the BBB to cortex and hippocampus at pharmacologically active concentrations, all without covalently modifying the ligand core. The hydrocarbon unit also repels intramolecular hydrogen bonding interactions between neighboring amide NH donors to ensure that the molecule adopts an extended conformation during transport, thus minimizing the cross-sectional area that must traverse the tight junction. On the other side of the barrier, the same linker acts as a membrane anchor that co-localizes the PROTAC with lipid-raft-associated E3 ligases, a subcellular preference that primes and speeds up ubiquitination of aggregation-prone substrates. Rodent pre-clinical data show that alkyl-linked degraders lower levels of pathological tau or α-synuclein species at oral doses that are an order of magnitude lower than for their PEGylated analogs, clearly indicating that the rate-limiting step was uptake, not catalytic efficiency. Since the alkyl chain can be radio-labelled at any carbon without changing polarity, the same scaffold can also double as a PET tracer for whole-brain occupancy studies to provide a frictionless translational bridge from rodent proof-of-concept to first-in-human doses. In summary, the hydrocarbon tether turns the alkyl linker from a simple spacer into a brain-penetrant delivery vehicle, a dual functionality that places it at the leading edge of next-gen CNS degradation campaigns.
Ordering and Support
At BOC Sciences, we streamline the process of sourcing hydrophobic alkyl linkers for research, discovery, and preclinical development. Our supply chain is built around speed, reliability, and analytical transparency, ensuring that every order meets the highest standards for reproducibility and purity.
Quality Standards and CoA Availability
Every batch of our alkyl linkers is delivered with a complete Certificate of Analysis (COA) and Quality Control (QC) documentation. Our analytical testing includes HPLC, LC-MS, and NMR to confirm molecular identity and purity. What you can expect from our quality assurance:
- Lot-to-lot c
- onsistency for reliable experimental results.
- Full spectral data for identity confirmation and traceability.
- Stability testing under controlled storage conditions.
- Detailed impurity profiling and residual solvent reporting.
These practices guarantee that every linker you receive is research-ready and validated for integration into your PROTAC or drug development workflows.
MOQ, Bulk Orders, and Custom Synthesis
We offer flexible supply options designed to meet the diverse needs of R&D and industrial partners:
- Minimum Order Quantity (MOQ): Starts at 100 mg for early-stage projects.
- Bulk Supply: Available in multi-gram to multi-kilogram quantities for scale-up or manufacturing support.
- Custom Synthesis: Tailor linker chain length (C3-C12), functional end groups (amine, azide, alkyne, NHS), or hybrid PEG-alkyl structures.
Our chemistry team is available to provide design assistance, helping you select or modify linkers for specific permeability, solubility, or binding profiles. We also support long-term supply agreements for biotech and pharmaceutical collaborations.
Partner with Us for Reliable Hydrophobic Alkyl Linkers
Hydrophobic alkyl linkers remain essential tools for improving membrane permeability and pharmacokinetic performance in modern PROTAC and small-molecule design. Choosing the right linker-and the right supplier-can directly impact your success in early discovery and preclinical optimization. At BOC Sciences, we combine analytical precision, scalable synthesis, and expert technical support to help you advance your programs faster and with greater confidence.
Contact our team today to request a quote, explore bulk options, or discuss a custom hydrophobic linker synthesis for your next drug development project. Let's accelerate your discovery with chemistry built for performance and reliability.
High-Purity Alkyl Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity Alkyl linkers.
FAQs
1. What makes hydrophobic alkyl linkers valuable in PROTACs?
They improve membrane permeability and compound stability, critical for drug development.
2. Are hydrophobic linkers suitable for CNS drug discovery?
Yes, their lipophilic nature enhances BBB penetration in central nervous system targets.
3. Can I customize the linker's chain length or functionality?
Yes, we offer C3-C12 linkers with optional end groups like amine, azide, or NHS.
4. What documentation is included?
All linkers ship with COA, QC reports, and full analytical data for validation.
References
- Zografou-Barredo N A, Hallatt A J, Goujon-Ricci J, et al. A beginner's guide to current synthetic linker strategies towards VHL-recruiting PROTACs[J]. Bioorganic & Medicinal Chemistry, 2023, 88: 117334. https://doi.org/10.1016/j.bmc.2023.117334.
- Troup R I, Fallan C, Baud M G J. Current strategies for the design of PROTAC linkers: a critical review[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 273. https://doi.org/10.37349/etat.2020.00018.
- Tedeschini T, Campara B, Grigoletto A, et al. Polyethylene glycol-based linkers as hydrophilicity reservoir for antibody-drug conjugates[J]. Journal of Controlled Release, 2021, 337: 431-447. https://doi.org/10.1016/j.jconrel.2021.07.041.
- Tashima T. Proteolysis-targeting chimera (PROTAC) delivery into the brain across the blood-brain barrier[J]. Antibodies, 2023, 12(3): 43. https://doi.org/10.3390/antib12030043.
- Distributed under Open Access license CC BY 4.0, without modification.
