Pomalidomide is one of the most illustrative cases of how such a simple imide core can be transformed into a versatile pharmacophore when its dual nature as an architecture with two chemically orthogonal domains (glutarimide for CRBN docking and phthalimide for exit-vector extrusion) is fully appreciated. The same modular design that confers nanomolar affinity to the ligase also provides well-defined, accessible handles in solution that can be extended, rigidified or decorated with non-carbon spacers without jeopardizing the pivotal hydrogen-bond trio. The drug is now a door-opener scaffold: in medicinal chemistry initiatives, the phthalimide plane is considered as an exchangeable springboard from which linkers transport additional recognition patterns toward any target-of-interest, while the glutarimide ring acts as an unchanged invariant key that anchors the whole construct to the ubiquitin system.
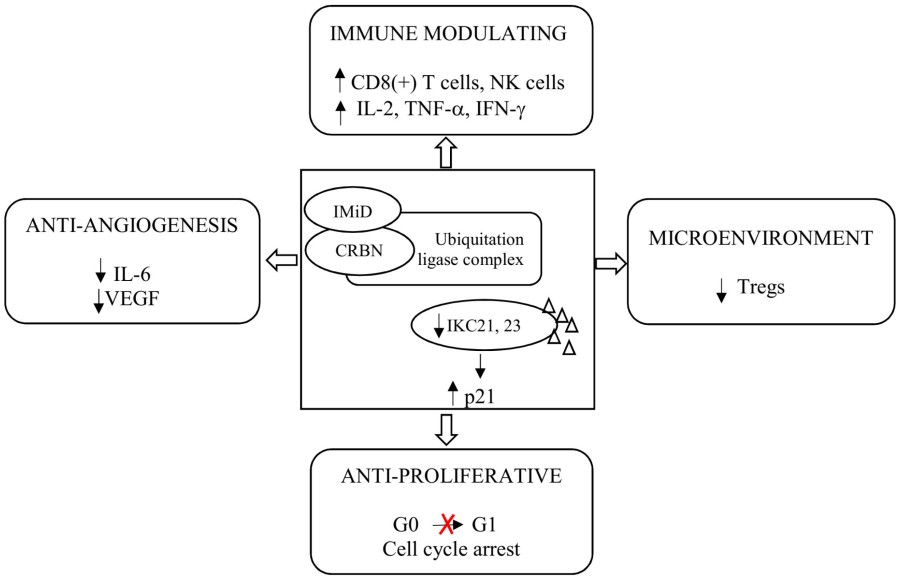 Fig. 1 Mechanism of action of immunomodulatory drugs.1,2
Fig. 1 Mechanism of action of immunomodulatory drugs.1,2
Core Structure and Chemical Properties
The pomalidomide scaffold may be thought of as a pair of cyclic imides that complement but do not overlap in electronic character. The glutarimide moiety is electron-rich, moderately acidic and conformationally flexible, which predisposes it towards reversible ion-pairing with CRBN's histidine-tryptophan pair. The phthalimide fragment is electron-poor, planar and hydrophobic, features that afford the glutarimide a steric shield against premature hydrolysis and an accessible π-face that may be functionalized without compromising the scaffold's bite angle for ternary complex assembly. This electronic polarity is the key to pomalidomide's chameleon-like solubility profile, in that it is lipophilic enough to traverse the lipid bilayer yet polar enough in its carbonyls to evade quantitative sequestration by serum albumins, a ratio which must be mirrored by linker engineering if the conjugate is to be cell permeable.
Glutarimide and Phthalimide Moieties Explained
The glutarimide ring is a pre-organized lactam-lactone hybrid with an internal dielectric that creates an enolic micro-environment at physiological pH. The two flanking carbonyls are hydrogen-bond acceptors that bind to backbone NHs in the thalidomide-binding pocket, and the imide NH donates a proton to an otherwise solvent-shielded CRBN carbonyl, forming a short, low-barrier hydrogen bond that is responsible for the slow off-rate that characterizes the series. As the ring is symmetric, it offers an identical electronic face to either side. This pseudo-C2 symmetry is broken once the phthalimide is fused at the 3-position, forcing the glutarimide into a half-chair that minimizes allylic strain and pre-organizes the lone pair trajectory required for nucleophilic attack during PROTAC synthesis. The phthalimide, for its part, is a rigid π-stacking platform that polarizes the adjoining imide carbonyl with its aromatic electrons, slightly lowering its LUMO and rendering the assembly more susceptible to mild nucleophilic aromatic substitution. This electronic activation is synthetically valuable: it permits late-stage installation of linkers under conditions compatible with acid-sensitive protecting groups, while simultaneously providing a spectroscopic handle (UV-active at 295 nm) that can be used to track conjugation efficiency by chromatography. Critically, the phthalimide plane sits at a dihedral angle of ~25° from the glutarimide mean, a vector that projects any appended linker away from the CRBN surface, reducing the entropic penalty encountered when the third partner protein is coerced into the nascent trimer. The two moieties thus work in concert: one for durable ligase capture, the other to provide a geometric and electronic springboard from which larger molecular edifices can be constructed without sacrificing the binding energy that is the glue mechanism's bedrock.
Key Physicochemical Parameters Affecting Reactivity
Pomalidomide is not particularly reactive, and its properties are less determined by overall polarity than by the spatial orientation of local partial charges, which are generated in the imide NH between an electron-withdrawing phthalimide and an electron-donating glutarimide. It has a narrow range of effective pKa around that imide NH; since it loses a proton at only a slightly higher pH than in vivo, a small decrease in local pH (as occurs, for example, in late endosomes) is enough to drive it into its anionic form. This form is more soluble, but also more prone to base-catalyzed hydrolysis. Its lipophilicity (used as a proxy for passive diffusion) is affected by an internal hydrogen-bonding pattern that effectively neutralizes the effect of its polar carbonyls; perturbation of this pattern by the attachment of a linker can increase its membrane affinity by a factor of 10, unless additional heteroatoms are added to the linker. Topological polar surface area (TPSA) is non-monotonic: a slight increase in TPSA relative to the parent value can improve oral availability, because the additional polarity disrupts a tendency to self-aggregate, which would otherwise form stable micelles in the stomach. The rotational entropy of the glutarimide ring is sensitive to steric protection; the addition of a single ortho substituent to the phthalimide can increase the energy barrier to ring rotation by several kcal/mol, which effectively rigidifies the molecule and increases residence time without changing the enthalpy of binding. The result of all this is that there is no simple one-to-one correspondence between global physicochemical properties and potency, and successful linker design must therefore walk a fine line between overall physicochemical properties and local conformational costs. This balancing act is now largely achieved with adaptive sampling algorithms, rather than rules of thumb borrowed from kinase inhibitor design.
Functionalization Sites and Reactivity
In the case of pomalidomide, the substitution patterns on C4 and C5 of the phthalimide are thus the primary offramp for structural diversity: they are electronically activated by the adjacent imide carbonyls, sterically available to solvents, and sufficiently removed from the glutarimide pharmacophore that electronic conjugation does not perturb the CRBN-binding triad. Modification at these centers is thus an electronic rheostat, tempering not only synthetic feasibility but also the rotational entropy of the neighboring aromatic moiety, the volume of the neo-substrate pocket, and ultimately the kinetic "on-off" ratio that determines which protein substrate is selected for ubiquitination.
C4 and C5 Modification Pathways
The electrophilic aromatic substitution reactions in mildly Lewis-acidic media direct substitution at C5 (para to strong electron-withdrawing carbonyl groups), enabling nitration, bromination or sulfonation at sub-ambient temperatures to form mono-substituted intermediates which undergo further modification through palladium-catalyzed cross couplings. C4 (meta to both carbonyls) is less activated but becomes the kinetic site when chelating substituents such as boronic acids are transiently directed by magnesium salts; this ortho-meta relay allows for sequential, regio-defined installation of two different side chains. Alternatively, oxidation of the phthalimide to a transient anhydride opens a protecting-group-free route: nucleophilic ring opening with amino alcohols installs a handle that can cyclize back to a substituted imide once the desired spacer is appended. Photoredox catalysis expands the toolkit further: excited-state iridium complexes abstract a single electron from the phthalimide, generating a radical cation whose spin density is highest at C4/C5; interception by cyano- or azido-containing radical traps delivers nitriles or azides that serve as click-ready linchpins. When applying mild protocols during late-stage processes scientists avoid the necessity of strongly acidic media which helps maintain the stability of linkers that are sensitive to bases as well as peptide fragments connected to the glutarimide side chain. Collectively, the synthetic menu now encompasses temperature-, redox- and metal-controlled pathways that convert the erstwhile inert aromatic plane into a versatile tapestry of electrophiles, nucleophiles and radical precursors without ever displacing the glutarimide gatekeeper.
How Substitution Influences CRBN Binding?
The crystal structures of substituted analogues show that C4 substituents point toward the solvent-filled channel between β-hairpin sensor loop while C5 substituents point into a hydrophobic pocket formed between two tryptophan "shelves". Small lipophilic groups on C5 make additional van-der-Waals interactions that prolong residence time and bias the loop toward the conformation that traps transcription factors with short aliphatic side chains. In contrast, polar or H-bond-capable groups on C4 are electronic signposts that reorganize interstitial water molecules that indirectly open the pocket and trap larger neo-substrates, including helical transcription factors. The structural impact is small, but critical: an amino group on C4 twists the phthalimide ring plane about ten degrees, which is enough to shift the position of the leaving linker by nearly an angstrom and change specificity from one family of zinc-fingers to another. Halogens on either carbon make a σ-hole interaction with a backbone carbonyl oxygen that acts as an enthalpic bonus, slowing off rates without affecting on rates. This level of regio-control was somewhat unexpected, as dual-occupancy of C4 and C5 does not universally result in an additive affinity: steric repulsion between a C5 methyl group and the C4 morpholine forces the aromatic ring into a high-energy twisted conformation, that transmits strain toward the glutarimide moiety, sometimes breaking the His-Trp clamp, and counterintuitively, causing weaker binding. In this manner, regio-chemical decision-making also serves as a conformational tuner: careful mono-substitution can improve both affinity and selectivity, while non-selective double substitution risks an entropic penalty that subverts the glue mechanism the scaffold was designed to leverage.
Synthetic Strategies and Practical Considerations
Scaling the pomalidomide scaffold to gram quantities for PROTAC or molecular-glue libraries is a balancing act of late-stage functionalization, protecting-group-free strategies, and impurity forensics: For effective gram-scale production of the pomalidomide scaffold in PROTAC or molecular-glue libraries scientists need free glutarimide nitrogen for CRBN binding and orthogonal handles on the phthalimide for linker attachment while implementing aqueous work-ups to prevent chromatography from becoming a production bottleneck. In practice, this translates to successful methods prioritizing convergent annulation, crystalline intermediates, and solvent swaps that minimize epimerization of the stereogenic glutarimide carbon—an apparently innocuous epimerate that completely abolishes the His-Trp clamp and seeds intractable diastereomeric mixtures downstream.
Common Synthetic Routes and Protecting Group Schemes
The most common approach to synthesis begins with the condensation of 3-nitrophthalic anhydride and 3-aminopiperidine-2,6-dione hydrochloride in refluxing acetic acid buffered with sodium acetate; under these weakly acidic conditions, the imide is formed in one pot, while the nitro group is preserved for further diversification. Reduction to the key 4-amino handle is most often accomplished by palladium-on-carbon hydrogenation at atmospheric pressure—an operation that is compatible with the glutarimide carbonyls but that does not result in the over-reduction that platinic catalysts will undergo. If orthogonal exit vectors are desired, the aniline nitrogen can be temporarily protected as a trifluoroacetamide, a group that withstands SNAr or acylation reactions on the phthalimide ring and can later be removed by mild carbonate hydrolysis, thereby releasing the reactive aniline without affecting the imide framework. If C5 functionalization is desired before the nitro reduction, a single-electron-transfer bromination under visible-light photoredox conditions installs a halogen that can be used in subsequent Suzuki or Sonogashira couplings; the trifluoroacetamide again acts as an internal base that suppresses a competing N-arylation. For linker attachment, aniline is most frequently linked via acid-chloride acylation in THF at reflux. This procedure provides the amide in a matter of hours, and the same step can be performed catalytically with 1-hydroxybenzotriazole and a water-soluble carbodiimide where acid-labile groups are present on the spacer already. During the route, crystallization is used in place of column chromatography wherever possible—ethyl acetate–hexane antisolvent trituration effectively washes away unreacted anhydride, and the product itself is typically isolated as a MeOH solvate that can be desolvated under vacuum without polymorphic drift. These choices—acetate-buffered condensation, hydrogenative nitro reduction, trifluoroacetamide protection, and crystallization-based purification—establish a protecting-group strategy that is both robust and telescopable, permitting multigram quantities of pomalidomide intermediates to be synthesized in two days in the laboratory.
Tips for High-Yield, High-Purity Derivative Production
To ensure reproducible yields greater than the critical amount required for downstream bioconjugation applications, three procedure details should be taken to obsessive lengths. First, the imide cyclisation reaction should be driven to complete conversion prior to any aqueous work-up, as residual anhydride trapped inside the precipitating imide will hydrolyse upon aqueous work-up, seeding colloidal acids that co-elute with the desired product and suppress subsequent amide coupling reactions. This can be monitored by a simple color test, whereby a basified aliquot of the imide is coupled with ninhydrin, as the absence of free amine is confirmed within minutes, thus negating the need for interim TLC testing. Second, the hydrogenation over palladium should be performed in a THF–DMF mixture rather than in neat alcohol, as the co-solvent prevents over-cooling during exothermic uptake and suppresses crystallization of the product on the catalyst surface which poisons the active sites and results in longer reaction times. Third, the final amide formation proceeds to much higher levels of conversion if performed using inverse addition, whereby the acid chloride is dissolved in iced THF followed by dropwise addition of the aniline solution; this keeps the concentration of free amine low and minimizes bis-acylation, ensuring that the only observable impurity is the easily rejected N,N-diacid. Removal of any colloidal palladium or aluminium salts on a short pad of neutral alumina prior to concentration prevents catalyzed hydrolysis during storage, and final drying under high vacuum at a temperature just below the onset of sintering affords a free-flowing powder whose residual solvent content is low enough to preclude hydrate formation, a common source of batch-to-batch variability in subsequent linker installation.
The interconnecting stretch between the CRBN ligand and the distal warhead is not simply a passive spacer, but a conformational spring whose length, torsional freedom and polarity influence the lifetime, geometry and sub-cellular distribution of the ternary complex. Successful optimization therefore proceeds by iterative negotiation between the enthalpic tightening of the E3–linker–POI triad and the entropic cost of freezing rotors that are otherwise beneficial for oral uptake. The most robust protocols now treat the linker as a programmable variable—sampled in silico, ranked by shape-embedding algorithms and translated into parallel micro-scale syntheses—rather than a static appendage appended after the pharmacophores have been fixed.
Choosing Linker Length, Rigidity, and Polarity
Early spongins featured alkyl or oligo-ethylene glycol linkers that were designed with the sole purpose of minimizing steric clash between the two ligands. Modern approaches take into account the observation that each additional rotatable unit increases the statistical ensemble of possible conformers, which decreases the population of productive binding poses and therefore increases the apparent concentration required for activity. This problem can be mitigated by shortening the linker to below a critical length, but this in turn forces the POI and ligase into strained, high-energy conformations that are accessible to kinetic sampling but not thermodynamic sampling. This is manifested by the compound having a rapid association rate but an equally rapid dissociation rate. In order to gradually increase rigidity, alkynes or E-alkenes were found to reduce torsional entropy without incurring the synthetic cost of fully bicyclic scaffolds, and para-substituted aromatics serve as planar hinges whose π-surfaces can engage in edge-to-face contacts with either protein, thereby adding favorable van-der-Waals interactions that compensate for the entropic penalty. Polarity must be similarly optimized in a parallel process: hydrophobic linkers tend to form micelles or bind to albumin, thereby decreasing the unbound fraction, while overly polar linkers can hinder translocation across lipid bilayers. The solution is often to incorporate a single, ionizable center, such as morpholine, a tertiary amine or a sulfonamide, in such a way that the pKa is at least one pH unit below physiological pH so as to ensure that the linker is only protonated (and therefore only soluble) in acidifying endosomes, after which the neutral species is restored in the cytosolic pH. Finally, modern, shape-directed algorithms can sample ensembles of virtual linkers whose end-to-end distances are in agreement with crystallographically observed separations. The top-scoring candidates are then filtered based on rotatable-bond count, synthetic accessibility and predicted metabolite stability, and the resulting shortlist is evaluated in parallel rather than via serial intuition.
Balancing Bioavailability and Binding Efficiency
Recall that these considerations form a continuum: the stiff, condensed linker that locks ternary affinity also packs into a crystal lattice that precludes aqueous solubility at an absorptive-qualifying level. Swapping the charge on one heteroatom could reclaim solubility but alter the recognized dipole moment of the linker; the purpose is therefore erased. A pragmatic alternative is to begin with an acceptance that perfect affinity is of secondary importance to catalytic turnover; provided residence time is on the minutes-scale or greater, further tightening has diminishing returns but even small improvements in bioavailability result in lower therapeutic doses and off-target load. Techniques include pre-metabolizing the linker in silico (soft esters or oxidative hot-spots that break to more polar fragments only after cellular entry) thereby retaining membrane permeability but allowing renal clearance of final products; or chimeric rigidity, a short alkynyl-stiffened section coupled to a flexible, polarity-tuned appendage that allows the central piece to meet geometric constraints while the terminal segment remains water-compatible enough to evade micelle sequestration. Parallel microsomal incubations and ternary-complex thermal-shift assays can provide a dual read-out that surfaces linkers which degrade faster than they precipitate, a signature that degradation has been traded-off for metabolic lability. Iteration continues until a Pareto frontier is approached: no further improvement in solubility is possible without structural modifications that impair complex stability, and vice versa. The resultant hit is therefore not an optimized parameter but a negotiated settlement embodying the acknowledgement that successful targeted degradation is a systems property of which PK and molecular recognition are inseparable facets of the same design space.
Analytical and Quality Control Techniques
Ensuring batch-to-batch equivalence of biological behavior for pomalidomide is anchored by orthogonal analytical sign-offs: chromatographic separation of the regio-isomeric linker attachments; mass-spectrometric confirmation that epimerization of the glutarimide ring has not occurred during final deprotection; and stress-testing of the candidate under humid, oxidative and photolytic regimens that approximate long-term storage conditions. The resulting data package is no longer a post-synthetic formality; it feeds back into synthetic planning, including the choice of solvent, temperature and even work-up sequence so that the final solid is crystalline, free-flowing and devoid of trace metals that could catalyze hydrolysis once the vial is opened.
HPLC and LC–MS for Structural Validation
Silica-based reversed-phase columns modified with covalently bonded polar functionalities provide baseline separation of the desired conjugate from its de-amino or N-oxide species and do not require ion-pairing agents that damage the mass spectrometer. Gradient conditions are often designed to begin with an acidic aqueous mobile phase to ensure silanols are protonated and do not cause tailing of the relatively planar phthalimide, followed by an increase in an organic solvent modifier of moderate polarity to elute late-running diastereomeric forms of the linker that are retained by conformational lock instead of polarity. High-resolution mass spectrometry acquisition in a combination of full-scan and data-dependent acquisition modes can simultaneously monitor the intact protonated parent ion as well as well as in-source fragment ions formed from opening of the glutarimide ring; the ratio of these two ions is a fast and simple diagnostic that the imide pharmacophore has not been lost in the final amidation step. If regio-isomeric functionality can occur, for example C4 versus C5 amination, coupling ion-mobility separation adds another gas-phase fractionation step to the analysis to separate overlapping peaks that have the same m/z but different collision cross-sections and thus avoids misinterpretation of the structural identity. System suitability is often checked by adding a mid-retention surrogate standard to ensure retention time variability is within method tolerance without reoptimizing the entire gradient.
Stability and Degradation Profiling
Forced-degradation experiments are therefore more about extending or exaggerating potential real-world stresses rather than driving total decomposition. Irradiation of samples in quartz tubes with cool white LEDs to simulate daylight exposure in ward packs, incubation in sealed dessicators at high humidity with saturated salt slurries, and repeated freeze–thaw to simulate clinical reconstitution are a few examples of these methods. In addition to measuring loss of potency, this strategy also requires monitoring the appearance of the specific hydrolytic products, the ring-opened glutarimide acids, des-amino phthalimides and linker cleaved fragments. The point at which these increase in level will give an indication of the most labile bond. Oxidative degradation from dilute hydrogen peroxide at near-neutral pH can produce N-oxides on the morpholine or tertiary amine functionality present in many new linker groups, which can then be purified by semi-preparative HPLC and the MS² spectra stored as 'fingerprints' for future routine analysis. Degradation in both the solid and dissolved forms also uncovers different pathways: the former being mostly surface hydrolysis limited by diffusion of moisture whereas the latter being autocatalytic following the introduction of even trace carboxylic acids that reduce the local pH. Mathematical modelling of these processes from the kinetics of these time-courses can generate algorithms that can predict shelf-life at ambient temperatures without requiring the slow real-time ageing and can help to inform potential reformulation changes (buffering excipients, crystalline to amorphous dispersions, nitrogen flushing etc.) earlier in development rather than after stability issues have been identified.
Our Technical Capabilities and Product Advantages
Expert Custom Synthesis of Pomalidomide-Based Reagents
We specialize in the design, synthesis, and optimization of pomalidomide-based reagents used in CRBN-mediated PROTAC development and molecular-glue research. Our chemistry team has extensive experience modifying pomalidomide at C4 and C5 positions, enabling precise linker installation without disrupting CRBN binding affinity. Through advanced synthetic route design, we ensure high yield, scalability, and structural fidelity across every batch. Whether you require functionalized pomalidomide intermediates, stable linkers, or ready-to-use PROTAC building blocks, our experts deliver compounds that meet the highest analytical and reproducibility standards. By combining synthetic innovation with deep knowledge of IMiD chemistry, we help researchers accelerate discovery and achieve consistent experimental success.
Comprehensive Analytical Testing and Documentation
Every pomalidomide derivative we produce undergoes multi-stage analytical validation to guarantee structural integrity, purity, and performance. Our in-house analytical suite includes HPLC, LC-MS, NMR, IR, and elemental analysis, supported by stability and degradation studies when required. Each product is shipped with a Certificate of Analysis (COA) and technical data package, providing full traceability for regulatory and publication purposes. We maintain strict batch-to-batch consistency, enabling researchers to compare data confidently across experiments and timepoints. Our commitment to rigorous quality control ensures your pomalidomide reagents perform precisely as intended -every time.
Competitive Pricing, Global Shipping, and Fast Turnaround
We understand that timing and cost are critical in research and development. Our optimized synthesis workflows and efficient logistics network allow us to offer competitive pricing, rapid production timelines, and worldwide delivery to academic, biotech, and pharmaceutical partners. We provide flexible order volumes - from milligram-scale research samples to multi-gram or kilogram-scale batches - supported by responsive technical and customer service teams. With a proven record of reliability, we make sourcing pomalidomide derivatives simple, cost-effective, and globally accessible.
Some Pomalidomide Derivatives We Provide
Partner with Trusted Experts in Pomalidomide Chemistry
Looking to design a new PROTAC or optimize your current pomalidomide derivative? Connect directly with our chemistry specialists to discuss your molecular design goals, synthetic feasibility, and analytical requirements. We provide personalized project consultation and help transform early-stage ideas into ready-to-use chemical solutions. Access our full catalog of high-purity pomalidomide intermediates and derivatives ready for PROTAC research. Request a quotation for research samples or bulk quantities, and our sales team will provide fast, transparent pricing and delivery timelines. All requests include access to technical datasheets and analytical documentation for easier evaluation and integration into your workflow. Request your quote now and start your next project with high-quality pomalidomide building blocks from a trusted global supplier.
FAQs
1. Which sites on pomalidomide are used for linker attachment?
The C4 and C5 positions are most commonly modified to preserve CRBN binding and allow diverse linker chemistries.
2. How does linker design affect PROTAC activity?
Linker length, rigidity, and polarity directly influence ternary complex formation and degradation efficiency.
3. What analytical techniques confirm pomalidomide integrity?
HPLC, LC-MS, and NMR are routinely used to validate purity and structure of pomalidomide derivatives.
References
- Image retrieved from Figure 1 " Mechanism of action of immunomodulatory drugs.," Charliński G.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Charliński G, Vesole D H, Jurczyszyn A. Rapid progress in the use of immunomodulatory drugs and cereblon e3 ligase modulators in the treatment of multiple myeloma[J]. Cancers, 2021, 13(18): 4666. https://doi.org/10.3390/cancers13184666.
- Nadeem O, Ailawadhi S, Khouri J, et al. Management of adverse events associated with pomalidomide-based combinations in patients with relapsed/refractory multiple myeloma[J]. Cancers, 2024, 16(5): 1023. https://doi.org/10.3390/cancers16051023.
- Barankiewicz J, Salomon-Perzyński A, Misiewicz-Krzemińska I, et al. CRL4CRBN E3 ligase complex as a therapeutic target in multiple myeloma[J]. Cancers, 2022, 14(18): 4492. https://doi.org/10.3390/cancers14184492.
