Few drug discovery stories have been as cinematic as the redemption of thalidomide, from sedative-synonym to cornerstone of targeted protein degradation. Thalidomide's infamous teratogenic liabilities, which precipitated its withdrawal from the market more than half a century ago, were systematically dissected to identify the E3 ligase cereblon (CRBN) as a central node regulating its multifaceted biological activity. Stepwise dismantling of the scaffold, coupled with scaffold hopping and co-crystal structure analyses, converted the parent molecule into a tunable handle that recruits neosubstrates for ligand-directed protein degradation. These discoveries positioned CRBN ligands as a modular Lego-brick for engineering heterobifunctional degraders, PROTACs, and molecular glues, where the focus of design strategies has since shifted from ligand-intrinsic pharmacology to engineered pharmacokinetics. We recapitulate the key steps along this journey that turned a parable into a paradigm.
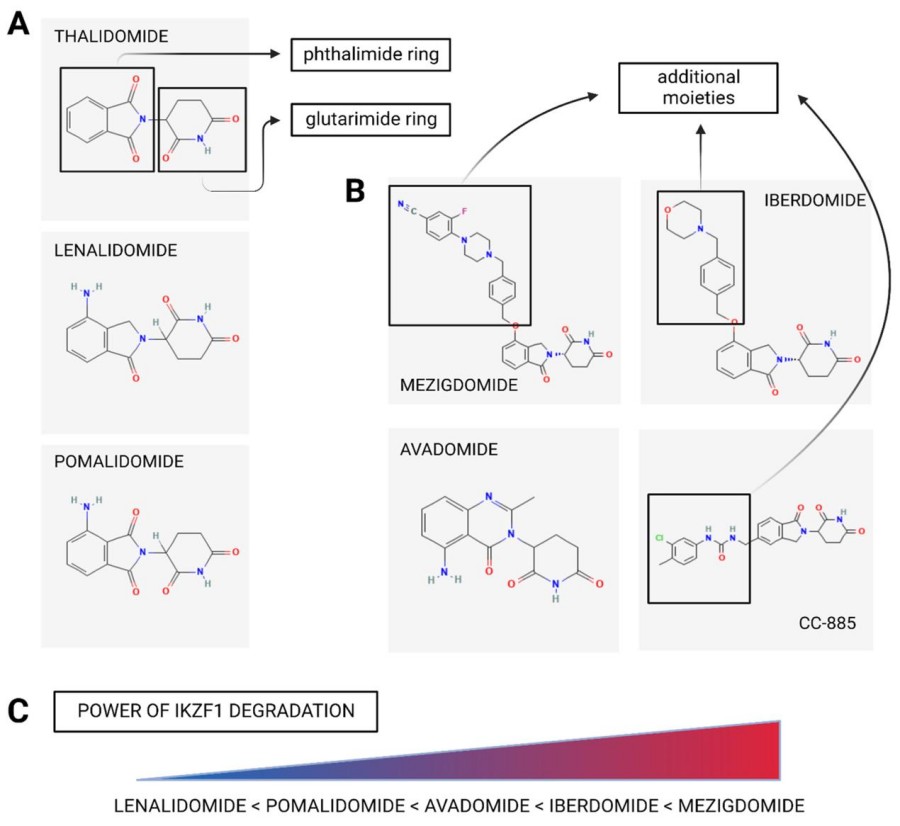 Fig. 1 IMIDs/CELMoDs differences in chemical structure and power of IKZF1 degradation.1,2
Fig. 1 IMIDs/CELMoDs differences in chemical structure and power of IKZF1 degradation.1,2
The Origins of Thalidomide and Its Discovery
Originally developed as a non-barbiturate hypnotic in the late 1950s, thalidomide was introduced to the clinic with the mistaken belief that it had an infinite therapeutic index, only to be withdrawn from the market worldwide as a result of the phocomelia epidemic. The consequences of this disaster were profound for drug safety and regulation, but the modern pharmacological investigation of thalidomide has been fuelled by a curiosity dating back to that time: how can an otherwise simple glutarimide core have both sedative and teratogenic effects? In other words, could thalidomide have unknown promiscuity? Reported off-label use of thalidomide over many years for leprosy and myeloma patients suggested anecdotal anti-inflammatory and anti-tumour activity and with this a cautious return to the laboratory. What followed was the description of a compound whose lipophilic core allows cell entry with surprising promiscuity to engage a substrate-recruiting element of CRL4 ubiquitin ligase, not as an expected antagonist but as a chemical switch. In this way, thalidomide's history and origin are tied to a demonstration of how limited target-agnostic drug development can be while at the same time providing a paradigm for ligand-dependent protein degradation that remains a key driver in medicinal chemistry today.
A Historical Overview: From Sedative to Research Tool
Thalidomide's renaissance can be divided into three periods of use that can be distinguished by their predominant user group and the identity assigned to the molecule. During the first period clinicians who were faced with the challenge of treating erythema nodosum leprosum noticed a reduction in the number and intensity of inflammatory relapses with thalidomide. This was ascribed to peripheral vasculature stabilization. Although their observations were anecdotal, they legitimized reintroduction in controlled clinical trials and they provided an epidemiological foundation for medicinal chemists, who in the second period began to dissect the phthaloyl-glutarimide scaffold in an attempt to develop molecules with improved risk–benefit ratios. The second period was defined when cell biologists recognized that thalidomide inhibits angiogenesis in rabbit cornea assays, and the insight that its teratogenic effects and antitumor activity might be two sides of the same coin, mechanistically rooted in cytokine modulation. This speculation, in turn, prompted further systematic modifications – an oxygen here, an amine there – and eventually led to the development of lenalidomide and pomalidomide without knowledge of the molecular target. The third period, which can be regarded as the transformative one, began with the discovery in 2010 that thalidomide binds to cereblon, a protein that converts the imide ring into a molecular handle that glues neosubstrates onto the surface of an E3 ligase. Thalidomide was no longer simply a rescued drug. Armed with mechanistic insights it became a chemical tool with which formerly undruggable signalling nodes could be targeted. The subsequent structural characterization of CRBN–ligand complexes has produced atomic blueprints, which are now being used for the de novo design of bifunctional degraders, PROTAC linkers and photopharmacological switches. So the story of thalidomide's rise from sedative to research tool is less a story of redemption than an example of how mechanistic understanding can transmute even the most inauspicious molecule into a cornerstone of modern target-validation platforms.
The Discovery of CRBN as the Thalidomide Target
Identification of cereblon as the drug target of thalidomide was based on three strands of evidence from the fields of chemical proteomics, development and enzymology. First, protein-capture experiments in which immobilized thalidomide derivatives were used as baits repeatedly identified a 50 kDa protein of unknown identity, however the relevance of this interaction was unclear until zebrafish embryos lacking the corresponding ortholog (crb) were found to exhibit tail fin- fold malformations that recapitulate thalidomide limb shortening phenotypes. Second, the finding that human CRBN already forms a complex with DDB1, CUL4 and RBX1, placed the candidate target in the middle of an E3 ubiquitin ligase complex, thus immediately raising the possibility that thalidomide might hijack rather than block ubiquitylation. This idea was substantiated through reciprocal pull-down assays which showed that the drug occupancy locks CRBN and CRL4 into a stable ternary complex with lineage restricted zinc finger transcription factors, thus targeting them for proteasomal degradation. Third, single amino acid substitutions in the thalidomide-binding pocket were shown to abrogate both thalidomide teratogenicity and anti-myeloma activity in mice, thus formally validating CRBN as the non-redundant mediator of all imide-based activity. Following resolution of the CRBN–ligand complex structure by X-ray crystallography, it became possible to use the three-dimensional architecture of the interface as a blueprint for the chemical grafting of exit vectors onto the glutarimide scaffold, thus giving rise to a family of hybrid molecules that like thalidomide strongly engage CRBN, but divert its ligase activity to a protein of choice. Thus, the discovery of CRBN did not only complete an outstanding mechanistic puzzle; it created an avenue that can now be used to harness the entire UPS for ligand-directed protein degradation.
Evolution of the IMiD Family
The IMDIs were not designed from a single rational design, rather they were derived from iterative rescue chemistry that gradually disassociated the desired immunologic effects from the sedative parent structure. Each iteration maintained the glutarimide "handle" necessary for CRBN binding, but re-sketched the periphery of the molecule so that neosubstrate engagement became tighter, more effective and - importantly - less toxic to embryonic tissue. In this way the family tree chronicles a gradual learning process in which medicinal chemists taught an otherwise promiscuous core to discriminate between myeloma cells and the transcription factors that control limb formation.
Lenalidomide and Pomalidomide as Safer Analogs
Informally, a heuristic of this evolution has been "keep the glue, lose the baggage". By removing one carbonyl from the phthaloyl moiety and appending an amino group at the 4-position, the conformational space was flattened: this both decreased the energetic cost to CRBN binding, but also reduced the plasma half-life. The result was an important decrease in neurotoxic potential and, surprisingly, it also provided the new derivative with the ability to target the zinc-finger protein CK1α for degradation—an effect not seen for the parent compound. Pomalidomide made a further play of the same logic, which appended an additional amino substituent to deepen the electrostatic interaction with a tryptophan shelf on CRBN. The additional hydrogen bond enhanced the tightness of the ternary complex, such that myeloma cells already resistant to lenalidomide could also be coerced to ubiquitinate IKZF1 and IKZF3. Critically, the second- and third-generation analogs also have lower partition coefficients in the blood–brain barrier, which has helped abate the peripheral neuropathy that limited chronic administration. Embryonic stem-cell assays further highlighted that SALL4, the transcription factor most directly tied to limb truncation, is either spared or only transiently engaged at therapeutic exposures. In sum, "safety" was not obtained by obviating CRBN affinity, but rather by re-tuning the kinetics of neosubstrate capture such that anti-plasma-cell efficacy could be achieved before teratogenic thresholds were reached.
Key Structural Modifications and Pharmacological Improvements
Medicinal efforts have interrogated every ring atom not required for recognition by cereblon. Early campaigns focused on the glutarimide nitrogen, and attempts to acylate or alkylate it to retard hydrolytic ring opening; though the resulting analogues were more chemically stable, they were uniformly lost affinity for CRBN, which demonstrated that the cyclic imide needed to be ionizable at physiological pH. Attention was then turned to the phthalimide half, and the bioisosteric replacement of the benzene ring with either thiophene or cyclohexene altered the vector along which the leaving substituent points. Such an exchange modified the spatial arrangement between the drug scaffold and the β-hairpin of CRBN, and hence helped re-orientate the complex toward transcription factors previously inaccessible from IMiD-mediated binding. A more recent strategy has made use of fluorine or cyano groups at the 5- or 6-position, by virtue of orthogonal σ-hole interactions, to reinforce binding without contributing to the molecular weight. Independent efforts have also shortened the linker that links glutarimide to the aryl unit to give conformationally locked bicyclic cores that are not subject to racemisation, and which show better oral exposure. In addition to the drug itself, formulation scientists have co-crystallized certain analogues with derivatives of cyclodextrin, giving rise to amorphous solid dispersions that help circumvent the very poor aqueous solubility that is otherwise inherent to the class. Taken together, these advances have increased systemic durability, improved neosubstrate selectivity, and carved a pharmacological window where anti-tumor activity is achievable at circulating levels that remain below the threshold for embryonic signal perturbation.
Understanding CRBN Binding Mechanisms
Binding between IMiDs and the C-terminal thalidomide-binding domain of CRBN follows a 2-anchor logic in which the glutarimide ring provides a highly conserved trio of hydrogen bonds that snaps the ligand into a pre-formed pocket, while the distal aryl moiety parades substituents onto an adjacent, more adaptable surface that can be re-sculpted into a neo-substrate trap. The mechanism is less a static lock-and-key than an induced-fit choreography in which ligand binding stabilizes a β-hairpin sensor loop, transiently turning CRBN from a passive receptor into an active degron reader. Understanding this switch-like behavior has become central to reconciling how minor chemical edits can lead to starkly different degradation profiles and how point mutations at the loop periphery can confer drug resistance without abolishing ligand association.
IMiD–CRBN Molecular Recognition
Crystal structures of ternary complexes identify a bipartite recognition motif. The glutarimide carbonyls are hydrogen bonded by the backbone amides of a tryptophan-histidine pair that functions as a kinetic clamp. The imide nitrogen, in turn, donates a hydrogen bond to a solvent exposed apo-state carbonyl. This set of interactions is largely conserved between first- and second-generation compounds, accounting for the fact that removal or replacement of the imide ring invariably results in inactivity. In contrast, the phthalimide or isoindolinone moiety of the drug occupies the entrance to a lipophilic channel bordered by the β-hairpin sensor loop and the Lon-domain helix. The geometry here is more permissive; small substituents such as an amino, methoxy or cyano group can make auxiliary hydrogen bonds or π-stacking arrays that tilt the loop to enlarge or contract the neo-substrate pocket. Water mediated bridges also commonly lock these interactions in place, so that small shifts in pKa or torsional rigidity translate into altered thermodynamic signatures. Notably, the ligand does not directly contact the doomed protein; instead it acts as a molecular splint that pre-organizes patches of electrostatics and hydrophobicity on CRBN, reducing the entropic cost of neo-substrate binding. This second-order recruitment mechanism explains why unrelated proteins with only a short glycine-rich β-turn in common can be recruited for ubiquitination, and why enantiomers of the same compound can have different substrate profiles even though they bind almost identically well to the receptor.
Comparative Binding Affinity and Structural Adaptations
Benchmarking through comparative calorimetry and surface-plasmon resonance (SPR) experiments has illustrated that prolongation of the aromatic backbone, e.g. through the addition of a phenyl-cyano or morpholine group, increases residence time without significantly impacting the on-rate, suggesting that unbinding is rate-determining and thus amenable to medicinal chemistry optimization. The associated structural change is an expansion of the tryptophan tunnel and a downward rotation of the sensor loop; this latter movement is limited by a conserved proline that functions as a pivot point, so that mutations at this residue disproportionately affect bulky exit vectors. By contrast, shortening of the aryl group decreases dwell time but can increase substrate selectivity, as the shrunken pocket no longer fits the bigger degrons. Halogenation at the 5-position adds a weak σ-hole interaction with a backbone carbonyl, which is absent in the native system; this single interaction, however, converts into a modest increase in affinity but also steers the loop towards a conformation that is optimal for recruitment of transcription factors with small aliphatic side chains at the important β-hairpin position. These subtle structure–adaptation nuances are a reason why semi-quantitative potency rankings are not transferable between neo-substrate classes: the same chemical modification that strengthens binding to CRBN may at the same time alter the surface presented to the third protein partner, resulting in a net degradation profile that cannot be reliably predicted without ternary structural information.
From Therapy to Research Chemistry
The emergence of IMiDs from merely "morning sickness soother" or "erythema nodosum suppressant" into drug-like chemical probes highlights how a certain logic of knowing pivoted around deciphering their mechanism of action: when the molecular glue hypothesis became clear, the scaffold that had been used to stabilize chemically produced hyperimmune conditions is then re-functionalized as a programmable handle to redirect ubiquitin machinery. This reclassification did not only lead to the prolongation of a medicinal class of molecules. It is from this moment on that an experimental syntax takes root by which any surface-exposed degron-containing protein can potentially be guided toward the proteasome with a suitably decorated glutarimide. The result is that the limits between therapeutics and bench reagents have been erased, and the IMiD core is now found simultaneously on oncology wards and in crystallization trays both as cure and as question.
How IMiDs Became E3 Ligase Ligands?
In contrast to the latter field, the therapeutic breakthroughs of the thalidomide derivatives had been empirically based. The physicians prescribing these compounds could describe what these drugs did (anti-cytokine, anti-angiogenic effects) but were unable to explain how they were working. The paradigm shift came when a 50 kDa protein was repeatedly co-purified from affinity pulldowns using thalidomide or other analogues. This protein was ultimately identified as the substrate receptor of CRL4. In subsequent studies the glutarimide was shown to occupy the same groove that the cognate substrates bound to, while the aryl appendage sticks out away from the surface, and thus provides an interface for non-native binding partners to bind to. Since this "molecular glue" mechanism was established, the field has realized that the IMiD scaffold is not simply a therapeutic agent but a ligandized master key to the E3 ligase. Thus medicinal chemists set about streamlining the scaffold to its core pharmacophore, removing elements that were involved in the teratogenic side pathway, but retaining the two carbonyls necessary for the His-Trp clamp. The resulting minimalist compounds had retained nanomolar affinity, could be conjugated via the solvent-exposed linkers, and crucially no longer required regulatory approval for research purposes, enabling widespread use of CRBN-directed ubiquitination.
The Shift Toward Targeted Degradation Platforms
With E3 ligand validation in hand, the field quickly transitioned from single-target glues to generalizable degrader platforms. Heterobifunctional molecules were designed in which the CRBN ligand constitutes one pole, a linker imparts spatial freedom, and a target-recognizing warhead occupies the other. Early scaffolds demonstrated that the catalytic process of ubiquitination alleviates the necessity for high occupancy; brief formation of a ternary complex is sufficient to poly-ubiquitinate and degrade targets previously deemed undruggable. Further encouraged by these proof-of-concept studies, groups diversified the chemical space beyond the classical phthalimide-glutarimide pair. Saturated heterocycles, reversed imides and even achiral dihydrouracils were identified that maintain the hydrogen-bond triad but offer orthogonal exit vectors. The linker itself also advanced from rudimentary alkyl chains to conformationally constrained spacers whose length and rigidity are optimized to the predicted distance between ligase and target epitope. Notably, this platform logic is agnostic to disease area: the same CRBN recruiter degrading a haematopoietic transcription factor in myeloma cells can, in conjunction with an alternative warhead, erase an oncogenic kinase in solid tumors. In this way, the IMiD legacy was abstracted into a plug-and-play grammar of degradation, catalyzing a broader movement in which drug discovery is no longer synonymous with enzyme inhibition but rather encompasses the spatial choreography of protein fate.
Modern Developments and Future Trends
Recent CRBN-targeted research has developed along two highly-interconnected axes: selective ligands that disconnect drug action from toxicity and design algorithms that use the ligase as a ‘plug and play' module instead of an optimized target. The overlap of these two fields is erasing the traditional distinction between lead optimization and de-novo design: off-target teratogenic liabilities are being surgically removed with scaffold hops guided by in-silico models, and generative models are suggesting chemotypes no human medicinal chemist would have thought of, yet they still obey the spatial rules of ternary complex formation. The net result will likely be a new generation of CRBN ligands that are safer, quicker to design, and applicable to disease indications well beyond the hematologic realm where the field originated.
Non-Teratogenic CRBN Modulators
A primary molecular liability that has shadowed the resurgent scaffold, however, has been the structure's tendency to distort normal developmental programs, which has motivated an effort to dissect which pharmacophoric elements are truly responsible for the biological efficacy and which are molecular vestiges of a nonimmunologic origin. In this line of thinking, recent drug-discovery efforts have thus embraced an aggressive scaffold migration, replacing the flat phthalimide with non-aromatic bicyclic systems, bioisosteric thiazoles or even contorted indolones that maintain the hydrogen-bond triad that clamps to the His-Trp pair, but present no π-π stacking surface for SALL4 binding. Analogous work with the glutarimide ring (gem-dimethyl locking, O to S replacement, or a conversion to a six-membered caprolactam) is also able to maintain the negative charge mimicry that is key for CRBN binding, while dampening the pharmacophore's charged properties linked to limb malformations. Also informative have been assays using embryonic stem cells as a read-out to create a phenomic landscape of analogues that are ranked according to how closely they map to a prototypical teratogenic signature; those molecules that make it through this screen uniformly have lower levels of persistence in the head mesenchyme, which itself suggests that having a short half-life in the target tissue is itself a risk mitigator. Significantly, the immune regulatory and anticancer activities of these redesigned ligands are also unimpaired, which suggests that the therapeutic and adverse events of CRBN targeting are, at a mechanical level, separable. In sum, these efforts are slowly resulting in a progeny of ligase ligands that clinicians might be more willing to prescribe to a fertile patient population or in long-term autoimmune indications where chronic use is anticipated.
AI-Guided Discovery and Next-Gen Ligase Chemistry
AI is currently being applied to the CRBN field to transition it away from a bespoke, iterative art form into a generative science that can draw thousand-member libraries overnight while also predicting ternary complex stability, off-target liability and synthetic tractability. Structure-conditioned variational autoencoders can now digest crystallographic ensembles, electron-density maps and hydrogen-bond propensities to learn the latent topography of the thalidomide pocket; these models can be subsequently used to emit novel cores that respect the geometric constraints of the β-hairpin sensor loop yet also probe regions of chemical space that are rarely visited in medicinal intuition. Reinforcement loops can be used to reward molecules predicted to form stable glue interfaces with disease-relevant proteins, penalize chemotypes that overlap with known teratogenic pharmacophores, and bonus scaffolds whose retrosynthetic complexity is below a user-defined threshold. In addition to raw speed, the conceptual innovation is in the inversion of design—rather than asking whether an acquired library contains a CRBN ligand, the algorithm is instead tasked with inventing the minimal structure that would satisfy a user-specified degradation signature. At the same time, physics-aware docking campaigns can be used to evaluate the kinetic feasibility of ternary complex formation in silico, flagging candidates whose linker length or exit-vector angle would otherwise strain the neo-substrate into an entropically prohibitive conformation. Initial experimental validation of AI-generated fragments shows hit rates orders of magnitude greater than historical high-throughput campaigns, suggesting that the bottleneck is no longer in finding starting points but rather in choosing among an embarrassment of computationally generated riches. The next logical step will be to integrate active-learning loops, where each wet-lab measurement is returned to the model within hours, to further compress these iterative cycles from months to days and usher in an era where ligase chemistry can evolve at the pace of the disease biology that it is targeting.
Our IMiD Product Range and Research Services
Thalidomide, Lenalidomide, and Pomalidomide Derivatives in Stock
We supply a complete selection of IMiD compounds - Thalidomide, Lenalidomide, and Pomalidomide derivatives - engineered for CRBN-based PROTAC research and molecular-glue discovery. Each product is available in analytical and bulk grades, ensuring suitability for both academic studies and industrial R&D programs. All compounds are verified by HPLC, LC-MS, and NMR, achieving purity levels above 98%, and come with full documentation including Certificates of Analysis (COA) and Safety Data Sheets (SDS). Our catalog supports rapid access to well-characterized IMiD ligands so you can focus on designing and testing next-generation degradation tools. Whether you are replicating foundational CRBN studies or developing selective degraders, our ready-to-ship IMiD derivatives deliver reproducibility and performance you can trust.
Some IMiD compounds We Provide
Analytical Data Support and Custom Functionalization
Beyond standard catalog items, we offer custom IMiD modification and linker-installation services tailored to your research goals. Our chemistry team can introduce functional handles at C4 or C5 positions without compromising CRBN binding affinity, enabling versatile conjugation strategies for PROTAC and molecular-glue applications. Each custom synthesis is supported by comprehensive analytical data - including purity validation, spectral characterization, and stability assessment - to guarantee full traceability and batch consistency. From early concept molecules to complex derivatives, we deliver high-purity IMiD scaffolds optimized for your target system and experimental design.
Why Researchers Choose Us: Quality, Consistency, and Global Delivery?
- Unmatched Quality: All products meet strict analytical standards for identity, purity, and stability.
- Consistent Supply: Reliable global logistics ensure timely delivery for recurring projects.
- Technical Confidence: Our experienced chemists and QC specialists provide direct consultation on ligand selection and synthesis routes.
- Flexible Options: Small-scale trial samples, mid-range R&D quantities, and large-scale bulk supply available on request.
These advantages make us a preferred IMiD supplier for biotech companies, CROs, and academic institutions worldwide.
Collaborate with Experts in IMiD Chemistry and PROTAC Innovation
Access technical datasheets, COAs, and analytical results for any compound in our IMiD portfolio. Submit a quick quotation request to receive pricing, availability, and lead-time details directly from our support team. We guarantee a fast response and transparent project communication.
Looking to push beyond standard materials? Collaborate with our R&D chemists to design and synthesize custom IMiD analogs or PROTAC intermediates that meet your project's unique structural and biological requirements. We provide end-to-end support - from molecule design and synthesis to analytical verification and delivery. Partner with us to accelerate your discovery pipeline with precision-engineered IMiD derivatives and trusted chemical expertise.
References
- Image retrieved from Figure 1 " IMIDs/CELMoDs differences in chemical structure and power of IKZF1 degradation.," Barankiewicz J.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Barankiewicz J, Salomon-Perzyński A, Misiewicz-Krzemińska I, et al. CRL4CRBN E3 ligase complex as a therapeutic target in multiple myeloma[J]. Cancers, 2022, 14(18): 4492. https://doi.org/10.3390/cancers14184492.
- Piccolomo A, Schifone C P, Strafella V, et al. Immunomodulatory drugs in acute myeloid leukemia treatment[J]. Cancers, 2020, 12(9): 2528. https://doi.org/10.3390/cancers12092528.
- Guo H. Bumped pomalidomide-based PROTACs[J]. Communications Chemistry, 2024, 7(1): 41. https://doi.org/10.1038/s42004-024-01125-2.
