Pomalidomide has additional utility beyond serving as a static inhibitor. As an integral component of a PROTAC, pomalidomide becomes a molecular chaperone, recruiting the E3 ligase cereblon in the presence of an otherwise-unliganded protein-of-interest. The small-molecule portion of the PROTAC provides a hydrophobic side and a glutarimide arm that cereblon accommodates as a natural substrate, thus eliciting an allosteric gesture that generates a transient cavity on the ligase. This transient binding site then provides the opportunity for the nascent ternary complex to ubiquitinate the neo-substrate, marking it for proteasomal degradation in place of temporary degradation. Due to pomalidomide's relatively low binding affinity for cereblon, the ubiquitin transfer is followed by a fast release of the pomalidomide, allowing the same ligase to catalyze several rounds of degradation while keeping the intracellular PROTAC concentration low to maintain pharmacodynamic activity. The efficiency of this "catch-and-release" mechanism has since turned the once lowly immunomodulatory imide into a cornerstone of contemporary degrader design, allowing chemists to trade out recruiting motifs but keep a cereblon footprint with which they are familiar, thereby expediting iterative design processes while avoiding off-target effects that historically impaired earlier generation heterobifunctionals.
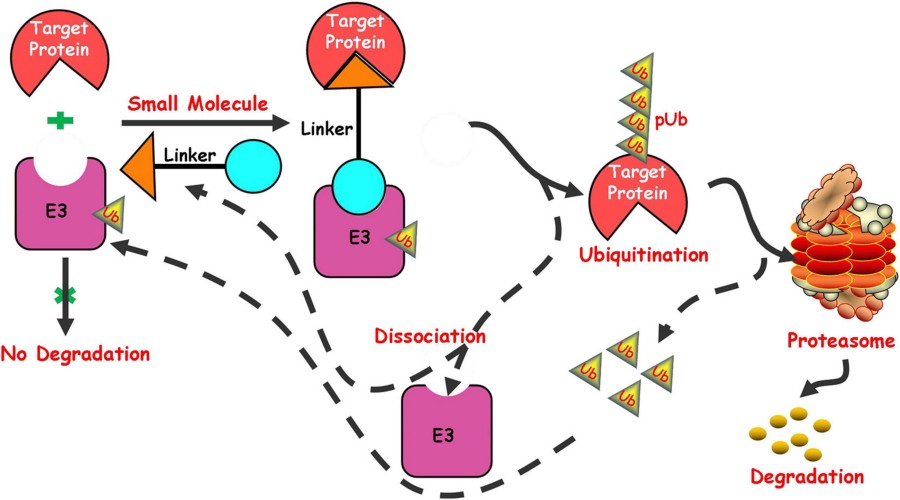 Fig. 1 Schematic depiction of the small molecule induced protein degradation.1,2
Fig. 1 Schematic depiction of the small molecule induced protein degradation.1,2
Introduction to Targeted Protein Degradation
Targeted protein degradation challenges our view of drug mechanisms, replacing one-time binding and a permanent "turn off" with the transient, event-based binding and reversibility of degradation. Rather than bind to an active site and alter an enzyme's kinetics, targeted protein degraders are designed to trick the cell's quality-control system into marking a target protein as misfolded or otherwise in excess, which is then subject to proteasome-mediated degradation until the protein is re-synthesized. For these reasons, this mechanism of action is largely resistant to traditional resistance mechanisms such as loss-of-occupancy point mutations, compensatory pathway activation, or feedback-mediated restoration of signal transduction after inhibitor dissociation. In addition, because degradation is a step downstream of enzyme function, it also overcomes target specificity issues for proteins that serve non-enzymatic, scaffolding or structural roles which cannot be targeted by orthosteric or allosteric inhibition. The story of a therapeutic shifts from "how strongly can we inhibit?" to "how specifically can we degrade?" In order to do this, molecules that both recognize the target with one moiety and an E3 ligase with the other must be designed to form a ternary complex that ultimately leads to ubiquitylation.
What Is PROTAC Technology?
PROTACs can be roughly thought of as a 'heterobifunctional' molecule that functions as a bridge between the protein-of-interest (POI) and an E3 ubiquitin ligase (LIG). PROTACs are most commonly composed of two pharmacophore modules linked by a spacer. Each module typically recognizes a unique binding site in its cognate protein, forming a ternary complex between the two proteins. In cells, after the PROTAC binds to both the POI and LIG, the ubiquitin ligase is brought into close proximity to the POI, which often induces ubiquitination of the POI. Successive rounds of ubiquitination can lead to the POI being targeted for proteasomal degradation. PROTACs are unique in that, after ubiquitination, the PROTAC dissociates from the ternary complex, allowing a PROTAC molecule to target multiple protein molecules for degradation (sub-stoichiometric activity). The discovery of a PROTAC for a given protein may involve in silico prediction of the protein–protein interface between the POI and LIG of interest, followed by screening of small molecule linkers to identify those that effectively bridge the two proteins. As such, this approach is also commonly known as 'linker-directed' discovery. The optimal linker length and geometry can vary, but are crucial for effective ubiquitination of the target protein. The requirement for a linker between the two moieties may provide opportunities to probe and target conformationally flexible binding sites that are deemed to be 'undruggable' by other methods. As the endpoint of PROTAC activity is degradation rather than occupancy, the intrinsic affinity required for PROTAC activity may be lower than for other small molecule targets.
Why Targeted Degradation Outperforms Traditional Inhibition?
Classical inhibition is like putting a solid lock on one door of a house. However, there may be side doors, tunnels, or a whole new gate can be constructed overnight by means of compensatory transcription. Targeted degradation, however, levels the entire house to the ground, so the cell needs to rebuild it brick by brick before signaling can be restored. This basic asymmetry gives rise to a number of pharmacological benefits. First, persistence: a single degrader molecule can undergo many catalytic cycles, so the pharmacodynamic effect can persist long after the plasma concentration has waned, which in turn eases the exposure–response curve and reduces peak-trough related toxicities. Second, specificity: as the formation of the ternary complex necessitates concurrent recognition of target and ligase, off-target binding events can usually abort before ubiquitination, constituting a built-in proofreading mechanism absent from occupancy-based drugs. Third, resistance resilience: point mutations that abrogate inhibitor binding also often disrupt protein fold and thus accidentally increase ligase recruitment and degradation rate—a perverse biological twist that uses the tumor's own escape mutation against itself. Fourth, broader target scope: scaffolding proteins, transcription factors, non-enzymatic structural hubs, that have no catalytic pocket to competitively block can now be eliminated en bloc, removing not only the enzymatic but also the architectural contributions to pathogenic signaling. Fifth, adjustable depth: by tuning linker geometry or ligase recruitment affinity, chemists can tune the fraction of protein removed, allowing graded knock-down for essential proteins that cannot be fully ablated. And last, but not least, degradation is downstream of allosteric regulation, which makes it indifferent to the complex feedback mechanisms that tend to reactivate pathway activity once inhibition is turned off. In the aggregate, this creates a mode of therapeutic engagement that functions less like an on/off switch and more like a rheostat with memory, which can continuously dampen oncogenic signaling while leaving overlapping, healthy circuits intact.
The Role of Pomalidomide in PROTAC Design
Pomalidomide has been repurposed from an IMiD into a recyclable adaptor which recruits E3 ligase cereblon without steric encumbrance at the opposite end of the chimeric molecule. Insertion of its glutarimide ring into a tryptophan-lined pocket flips the receptor into a "guest-welcoming" conformation that transiently accepts neo-substrates. This interaction is reversible, and occurs with rapid on/off kinetics, which allows the ligand to shuttle back to escort fresh targets to the ubiquitin machinery, permitting sub-stoichiometric dosing. Synthetic handles decorating the aromatic core are tolerant to nucleophilic aromatic substitution or metal-catalyzed cross-coupling, allowing chemists to tag a wide range of linker-payload combinations without perturbing the crucial CRBN interface. This feature set – validated ligase engagement, chemical modularity and clinical safety pedigree – has made pomalidomide a de facto CRBN anchor for first-generation PROTAC libraries, and into a benchmarking reference against which newer scaffolds are judged.
Understanding IMiD Compounds as CRBN Ligands
IMiDs are a class of glutarimide-containing small molecules which subvert the substrate specificity of the cereblon-DDB1-Cul4A-Rbx1 E3 ligase towards substrates it was not designed to ubiquitylate. The first, glutarimide, component binds to a tri-tryptophan binding pocket on cereblon, with the second, typically aromatic, moiety determining the neo-substrate which is recruited. This induces an allosteric effect which contracts a sensor loop, locking the ligase into a "closed" conformation and generating a new binding pocket in which to hold β-hairpin degrons found on IKZF1, IKZF3 or GSPT1, for example. This accounts for how IMiDs can alter degradation target by small structural changes (i.e., an amino group moved by one carbon or a carbonyl switched for an ether) without a change in CRBN affinity. The allosteric effect also accounts for how an endogenous cereblon function in glutamine metabolism is inhibited, which is an off-target event likely to the teratogenic effects of thalidomide. IMiDs are therefore, from a chemical biology perspective, not just ligands but reprogrammable adaptors which convert the small-molecule language of small-molecule recognition into the protein language of protein-protein proximity which is interpreted by the cell as "degrade now". Catalytic activity in a PROTAC is dependent on this reversibility: once polyubiquitin has been attached to the target, the IMiD can dissociate and come back to tag another protein. It is for this reason that the glutarimide core ring is considered a privileged warhead by medicinal chemists, whose key interactions are kept unchanged. Aromatic sidechains on the second moiety can be varied to modulate neo-substrate choice, linker angle and drug-like properties (solubility, metabolic stability, etc). The mechanistic understanding of IMiD behavior down to the atomic level has also helped answer questions around how and why certain cancer cell lines are sensitive to IMiD monotherapy, and resistant in others: it is less a question of CRBN levels but a question of neo-substrate degron availability and cell ubiquitin-proteasome flux. This knowledge is now used to aid incorporating IMiD modules into heterobifunctional degraders while ensuring the ligase-recruiting portion of the degrader remains in the correct reading frame.
Structural Benefits of Pomalidomide vs Other IMiDs
As such, pomalidomide straddles the thalidomide and lenalidomide design spaces: a design in which its 4-amino substituent on the phthaloyl ring is orthogonal in two ways. The additional hydrogen bond provided by the substituent has not sacrificed the rotational entropy cost of larger heterocycles, allowing for a tighter fit into cereblon's tryptophan cage without compromising its conformational flexibility. The upshot is faster association kinetics with a residence time long enough to allow for ubiquitin transfer but short enough to prevent steric sequestration of the ligase (lenalidomide can be too electrostatically complementary). In addition, the electron-rich aniline of the phthaloyl allows for late-stage functionalization via mild nucleophilic aromatic substitution under conditions that are orthogonal to ester, amide, or carbamate linkers, affording diverse analogues that thalidomide's unsubstituted core does not allow. Metabolically, pomalidomide also has an edge over thalidomide. Phthalimide hydrolysis is a known metabolic liability for the first-generation immunomodulators, and the amino group on pomalidomide reinforces the imide carbonyl against nucleophilic attack from ring-opening esterases. In terms of degradomics, the presence of the 4-amino substituent also slightly alters the neo-substrate landscape by favoring the degradation of transcription factors that have an acidic patch in close proximity to the mandatory β-hairpin degron. In other words, the targetable proteome is expanded beyond IKZF1/3 to include transcriptional co-repressors and splicing factors. Lastly, the safety profile of pomalidomide in a clinical setting is also well-established over its 10 years as a myeloma drug. In other words, regulators are already familiar with the cereblon-pomalidomide interaction and should be more accepting of risk when the scaffold is repurposed into PROTACs. These are the structural rationales for why pomalidomide is the default cereblon ligand for most proof-of-concept degraders and why newer and higher-affinity cereblon mimics are still benchmarked against.
Mechanism of Action - How Pomalidomide Recruits CRBN
Pomalidomide acts more as a conformational key than a classical ligand; it temporarily opens a previously inaccessible crypt in the cereblon E3 adaptor, with its glutarimide ring wedging between three highly conserved tryptophan residues to force a hinge-mediated closure that remodels the substrate-recognition platform. The allosteric tilt redefines, rather than simply increasing, the chemical grammar by which CRBN interrogates potential protein partners, thus allowing the ligase to misidentify neo-substrates as endogenous clients. The event is transient—pomalidomide's relatively low residence time ensures that the small molecule departs once ubiquitin has been added to the target, allowing CRBN to participate in further cycles of ubiquitination. Thus, pomalidomide acts more as a catalytic match-maker than a static occupant, a feature that reduces the intracellular dose needed for pharmacology and minimizes off-state sequestration of the ligase pool.
Step-by-Step View of the Ubiquitination Cascade
The pomalidomide saga commences as the drug slips through the lipid bilayer and stumbles into the CRBN-DDB1-Cul4-RBX1 holoenzyme. Within milliseconds, the glutarimide head docks into the tri-tryptophan cradle, simultaneously rotating an α-helix that functions as a molecular turnstile. This motion unveils a nascent hydrophobic patch on the upper surface of CRBN that, in effect, posts a "vacancy" sign for β-hairpin degrons from substrates such as IKZF1 or IKZF3. Neo-substrate arrival gives rise to a transient ternary complex. No covalent bond is formed, but the net binding energy of the ensemble is enough to bring a lysine side chain within 8–10 Å of the E2~ubiquitin thioester. Next, RBX1, the RING finger subunit of the E3, catalyzes a thioester hand-off: ubiquitin is transferred from the charged E2 to the exposed lysine in a reaction that proceeds at biologically relevant rates because an extended network of ordered water molecules shields otherwise repulsive charges. Once the first ubiquitin is grafted, the same lysine or neighboring residues are revisited in a processive manner, resulting in the growth of a K48-linked chain in a beads-on-a-string fashion. When chain length exceeds four ubiquitins, the 26S proteasome's regulatory particle recognizes the signal and engages the substrate through a combination of hydrophobic gripping and unfolding. ATP-dependent translocation threads the tagged protein into the proteolytic chamber where processive cleavage yields short peptides that are ultimately recycled into the amino-acid pool. Throughout this choreography, pomalidomide remains non-covalently bound; once the substrate is committed to degradation, thermal breathing of the CRBN pocket ejects the small molecule, allowing it to re-enter the cytosolic crowd and catalyse another cycle. The entire cascade—from initial encounter to proteasomal destruction—can conclude within minutes, which is why pharmacodynamic effects often precede measurable changes in transcript levels.
Ternary Complex Formation and Protein Elimination
The geometric details of ternary complex formation are the key to degradation success or failure. Crystal structures indicate that pomalidomide serves as a rigid linker to tether CRBN and neo-substrate in a V-shaped configuration rather than pressing the two surfaces together directly. This orientation reduces steric clashes between bulky patches while simultaneously orienting the target lysine towards the E2 catalytic groove. The linker exiting the phthaloyl ring is also solvent-accessible, and so represents a chemical "handle" that PROTAC designers can modify without disrupting the core trio of contacts. Charge-based complementarity also serves as a gate-keeping step: acidic residues on either side of the β-hairpin degron form salt bridges with basic patches on CRBN, imposing an additional layer of selectivity on top of the primary hydrophobic plug. These electrostatic contacts are distance-dependent, and so even a single methylene insertion into the linker will be sufficient to disrupt ternary complex stability, making sub-ångström precision during design paramount. Once the ternary complex has formed, the binding energies are often synergistic – or "cooperative" – so that the total avidity often exceeds the sum of individual affinities. In other words, weak monovalent interactions can still result in strong degradation as long as the geometry is favorable. CRBN melting temperatures, as determined by thermal shift assays, increase only slightly upon pomalidomide binding but increase sharply upon addition of the neo-substrate, which confirms that the ternary state is the most thermodynamically stable complex. Curiously, the same induced-fit motion that licenses ubiquitination also occludes a cereblon epitope that is recognized by endogenous inhibitors, serving as a biochemical "timer" that limits degradation to the brief time during which all three molecules are on stage at once.
Design and Optimization of Pomalidomide-Based PROTACs
Drug design for pomalidomide-based PROTACs is somewhat different from other ligands: pomalidomide is treated as a rigid hinge instead of a fixed point, with the phthalimide as the base which gives good affinity to CRBN, while the atoms surrounding the phthalimide are modified to give suitable chemical handles for linker conjugation, without disrupting the sensitive indole ring. Drug design is then further constrained by three main rules: 1) keeping nanomolar affinity for ternary complex formation, 2) keeping solubility under different ionic microenvironments, and 3) keeping instability after payload delivery, rather than before. Drug design therefore cycles through atomic-level electrostatic profiling of the C4/C5 substituents, and coarse-grain simulation of overall molecular shape, to find designs that act as catalytic spacers, rather than blunt-ended blockers. The evolving design principles in pomalidomide-based PROTACs therefore involve mid-sized linkers, with switchable flexibility: a stiff spacer close to the ligase, to lower the entropic cost, then a short alkyl chain that gives the target-facing warhead rotational liberty to find its target. Initial proof-of-principle libraries focused on kinases, epigenetic readers, and oncogenic GTPases, have shown that pomalidomide-based PROTACs can outperform parent inhibitors at sub-catalytic doses, setting the stage for further expansion of the platform to other therapeutic areas, beyond cancer, such as fibrosis, immunology, and neurodegeneration.
Key Modification Sites (C4, C5) and Their Chemical Behavior
The electronic gatekeeper at C4: the amino substituent is an electron-donating group into the phthalimide π-system. This strengthens the van-der-Waals interaction with the tri-tryptophan pocket and provides a nucleophilic handle for the introduction of amide, carbamate or urea linkers. Modifications at C4 have only a small effect on the torsion angle of the glutarimide ring, keeping the affinity for CRBN within a logarithmic scale of the parent ligand. When moving the substitution to the adjacent C5 position, there is room for bigger groups. Depending on the exit vector, small ethers or fluoroalkanes have minimal interference with the cereblon contacts while larger aromatic substituents induce a twist of the imide plane and weaken binding. Chemists leverage this structural toggle by using orthogonal protecting groups, allowing for late-stage functionalization under mild photoredox or palladium-free conditions to avoid racemization of chiral centers further down the linker. Regioselective nitration followed by reduction provides a C4-aniline that can be employed in Sonogashira or Buchwald–Hartwig couplings to introduce sp- or sp²-hybridised tethers affecting overall molecular rigidity. Moreover, electron-withdrawing groups at C5 can enhance metabolic hydrolysis of the glutarimide, which leads to degradation of the PROTAC. Therefore, electronic tuning has to be balanced with microsomal stability assays. C4 and C5 act as tunable resistors in this sense: C4 modulates conjugation chemistry while maintaining CRBN π-stacking and C5 influences lipophilicity and clearance, which opens up a two-dimensional space for optimization without backtracking on the key recognition motif.
Linker Considerations and Spatial Orientation
The identity of the linker determines whether the pomalidomide-CRBN encounter happens on the same time scale as target engagement or in a kinetically distinct step. If the tether is too short, it creates steric clashes that force the ternary complex to adopt a high-energy conformation that the ubiquitination machinery will not recognize; if the strand is too flexible, the binding event is entropically favored and off-target interactions will be more likely, reducing the drug's potency. For this reason, linkers are often composed of functional "stanzas": an N-terminal rigid unit (often an aryl or alkynyl group) that projects the molecule away from the ligase surface, a middle section that can hydrogen-bond with the solvent to improve solubility, and a C-terminal flexible unit that allows the warhead to sweep a spherical sector in search of its epitope. Metadynamics simulations indicate that a 12- to 16-atom distance places the target lysine residue within 8 Å of the E2 active site while still keeping the total entropic penalty below the threshold for ubiquitin transfer. Furthermore, the insertion of a single ether oxygen atom per four carbons creates a "kink" that prevents extended hydrophobic collapse and improves solubility without reducing membrane permeability. Orientation is also influenced by the number of rotatable bonds: having fewer freely spinning sigma bonds close to the phthalimide warhead reduces the entropic cost of ternary complex formation, while having some controlled flexibility near the warhead end can allow induced-fit adjustment to bind sub-optimally positioned lysine residues. Traceless linker cleavage motifs (carbonates or boronic esters) can also be introduced so that linker byproducts can evaporate after the protein is marked for degradation, reducing off-target effects caused by the scaffold itself.
Representative PROTAC Examples (BTK, HDAC, BRAF)
The PROTACs above also highlight another key aspect of early BTK-directed degraders: coupling pomalidomide to a linker-embedded acrylamide recognition motif using a 14-atom long alkyne-ether rod achieved sub-catalytic clearance of the kinase at circulating drug levels well below those necessary for traditional occupancy-based inhibition. The ternary crystal structure shows that the complex pre-organizes Lys430 into hydrogen-bonding distance of the incoming E2, allowing for processive ubiquitination despite modest intrinsic affinity. Shifting focus to HDAC isoforms, the optimization bottleneck moved from access to the catalytic cysteine to nuclear localization: introducing a basic amino-ester near the center of the linker backbone exploited transport by importin-α to shuttle the degrader into compartments where class-I deacetylases are enriched. Inside the nucleus, the flexible portion of the linker enabled the hydroxamic acid warhead to sample the catalytic tunnel, while the rigid segment held constant the distance and geometry between drug and CRBN to achieve selective HDAC3 degradation and downstream hyperacetylation of histones without pan-HDAC cytotoxicity. BRAF degraders had yet another hurdle to overcome: as the kinase flips between active and inactive states, each flip-state presents a different groove on its surface. A pomalidomide-based PROTAC with a bifurcated linker rigid for the DFG-in state but extendible for the DFG-out state 'captured' both conformers, resulting in pan-variant degradation and postponing the onset of resistance mutations which are commonly seen with ATP-competitive inhibitors. A common theme through all of these stories is that pomalidomide is a recurring narrator: while its affinity for CRBN is the foundation of the story, changing the linker-warhead 'paragraphs' allowed the script to be tailored to different oncogenic 'characters', demonstrating the platform's applicability to solid-tumor and immune indications beyond haematological malignancies.
Advantages and Common Challenges
Pomalidomide-based PROTACs are an important part of the degrader landscape: The boron heterocycle offers both exceptional potency in recruiting cereblon as well as a favorable safety profile that is further supported by a clinical legacy. However, these same properties also give rise to a number of inherent challenges. On the one hand, the glutarimide cap interacts with a highly preserved tri-tryptophan pocket and displays an off-rate that is fast enough to avoid an off-target pathology from irreversible ligase trapping, allowing for sub-stoichiometric turnover and reducing the risk of on-target accumulation. Moreover, the nucleophile-rich phthaloyl moiety allows for late-stage modification, letting researchers graft on different rigid or flexible linkers without the need to reoptimize interactions with CRBN afresh. On the other hand, this platform is highly promiscuous for various zinc-finger transcription factors, and as a result off-target binding and co-degradation is a common refrain: IKZF1, IKZF3 as well as a handful of unannotated C2H2 zinc fingers are often degraded along with the target, with immunosuppressive consequences that can obscure the sought-after pharmacological effect. The key creative struggle for the field thus becomes how to capitalize on CRBN-targeting potency while muting the noise of off-target zinc-finger degradation.
High CRBN Affinity and Biological Compatibility
The glutarimide ring of pomalidomide acts as a sort of pre-evolved skeleton key: it wedges between Trp380, Trp386 and Trp400 of human cereblon, triggering a hinge motion that snaps shut the substrate-recognition lid without displacing the ordered water networks involved in downstream ubiquitin transfer. The induced-fit event is rapid and reversible, allowing residence times long enough for poly-ubiquitin stamping but short enough to avoid the ligase "trapping" that's a pharmacological dead-end with higher-affinity heterocycles. Electron-donating substituents at the C4 aniline position deepen π-stacking into the indole face, progressively strengthening affinity without triggering the entropic penalties of macrocyclization. The ligand also does not oxidize the Cul4-Rbx1 catalytic core nor otherwise interfere with neddylation cycles required for ligase renewal, thereby sparing global ubiquitin homeostasis in non-transformed cells. Metabolically, the imide carbonyls are relatively poor substrates for amidases at physiological pH; instead, the molecule undergoes gentle phase-II glucuronidation that preserves oral bioavailability across species. Because CRBN expression levels are relatively consistent in most somatic tissues, pomalidomide-based PROTACs have a broad therapeutic index window, and prior clinical experience with the parent IMiD has produced a comprehensive safety lexicon that regulators will accept with minimal additional lexicography. In sum, these attributes give the modulator a favorable translational runway: teams can focus linker optimization on target engagement rather than re-engineering ligase compatibility, which shortens iterative cycles and frees up capital for downstream pharmacology studies.
Off-Target Zinc Finger Degradation and How to Minimize It
The same conformational "latch" that ensures binding of IKZF1/3 also senses a consensus β-hairpin degron present on many other C2H2 zinc-finger proteins, leading to off-target degradation of transcriptional guardians that control hematopoietic lineage commitment. The resulting lymphopenia can be misinterpreted as on-target efficacy and mask the presence of dose-limiting toxicities until late-stage clinical trials. In an effort to increase selectivity, researchers have begun implementing local steric filters: a short PEG-like insert located adjacent to the C4 aniline demands a larger hydrophobic "footprint" from the incoming zinc finger, sterically repelling smaller transcription factors that are unable to pay the entropic price. In another approach, the canonical flexible linker is swapped for a semi-rigid aryl-alkyne rod that directs the warhead away from the CRBN surface, such that only those targets with extended docking epitopes can bridge the gap. Electrostatic gating of the guest list is also being explored: incorporation of an anionic sulfate or carboxylate group on the linker shaft precludes binding to C2H2 domains that lack complementary basic residues, yet still permits binding to acidic pockets that are present in desired oncogenic clients. Finally, innovation in scheduling is being considered: pulsatile dosing regimens that allow for lymphocyte repopulation between exposure windows may mitigate cumulative immunosuppression while retaining tumour cell kill. By layering orthogonal filters (steric, geometric, electrostatic, and temporal), modern pomalidomide PROTACs progressively narrow the set of co-degraded zinc fingers and shift the therapeutic index from a liability-laden footnote to a controlled narrative variable.
Our Pomalidomide Derivatives and Custom PROTAC Solutions
High-Purity Pomalidomide Analogs for Research and Development
Our laboratory offers a comprehensive portfolio of pomalidomide analogs and IMiD-based derivatives specifically designed for CRBN-mediated PROTAC research. Each compound is synthesized and purified to high HPLC purity, ensuring consistent performance and reproducibility across both academic and industrial studies. We provide multiple grades - from research-use quantities for discovery projects to bulk lots suitable for preclinical screening or scale-up chemistry. All batches include full LC-MS, NMR, and COA documentation, enabling seamless integration into your analytical and regulatory workflows. Whether you are developing oncology-focused degraders or exploring cereblon biology, our high-purity pomalidomide ligands serve as the reliable foundation for efficient and selective PROTAC design.
Tailored Linker Attachment and Derivative Design
Beyond catalog compounds, we specialize in custom pomalidomide functionalization and linker conjugation. Our expert chemists can modify the molecule at C4 or C5 positions, preserving optimal CRBN affinity while introducing functional handles for diverse linker architectures - such as alkyl, PEG, or aromatic spacers. We work closely with research teams to design linkers that achieve the right balance of flexibility, polarity, and length, ensuring effective ternary complex formation between the target protein and cereblon. If you're optimizing degradation potency or minimizing off-target zinc finger effects, our structure-activity-driven synthesis approach delivers data-backed results. With custom linker design and derivative engineering, we transform your conceptual PROTAC scaffold into a ready-to-test research compound.
Advantages: Quality, Speed, and Technical Expertise
- Uncompromising Quality: Every batch is verified by HPLC and LC-MS to guarantee structural integrity and high CRBN-binding potential.
- Fast Delivery: Streamlined synthesis and logistics enable rapid turnaround from inquiry to shipment.
- Experienced Scientists: Our chemistry team combines expertise in IMiD chemistry, linker engineering, and PROTAC optimization.
- Collaborative Support: Direct access to R&D chemists for design feedback and synthetic feasibility evaluation.
These strengths make us a trusted global partner for PROTAC discovery, offering precision chemistry and dependable research materials.
Some Pomalidomide Derivatives We Provide
Collaborate with Experts in Pomalidomide and PROTAC Chemistry
Get immediate access to technical datasheets, COAs, and spectral data for any pomalidomide derivative in our catalog. Submit a quotation request to receive customized pricing and lead-time estimates for research, pilot, or bulk-scale orders. Our support team responds within 24 hours to ensure your project moves forward without delay.
Looking for more than standard materials? Collaborate directly with our scientific development team to co-design, synthesize, and optimize custom PROTAC reagents based on your target or E3 ligase strategy. We offer end-to-end support - from ligand derivatization to linker optimization and analytical validation. Partner with us to accelerate your next-generation PROTAC discovery using tailored pomalidomide chemistry and proven manufacturing expertise.
FAQs
1. How does pomalidomide contribute to targeted protein degradation?
Pomalidomide recruits the E3 ligase cereblon (CRBN) to the target protein, enabling ubiquitination and proteasomal degradation through PROTAC mechanisms.
2. Why is pomalidomide preferred over other IMiDs in PROTAC design?
It offers stronger CRBN affinity, greater linker modification flexibility, and higher degradation selectivity compared to thalidomide or lenalidomide.
3. What are common applications of pomalidomide-based PROTACs?
They're used in oncology, immunology, and neurodegenerative disease research to remove difficult-to-drug proteins efficiently.
References
- Image retrieved from Figure 1 " Schematic depiction of the small molecule induced protein degradation.," Li X.; et al., used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy[J]. Journal of hematology & oncology, 2020, 13(1): 50. https://doi.org/10.1186/s13045-020-00885-3.
- Liu Z, Hu M, Yang Y, et al. An overview of PROTACs: a promising drug discovery paradigm[J]. Molecular biomedicine, 2022, 3(1): 46. https://doi.org/10.1186/s43556-022-00112-0.
- Qi S M, Dong J, Xu Z Y, et al. PROTAC: an effective targeted protein degradation strategy for cancer therapy[J]. Frontiers in pharmacology, 2021, 12: 692574. https://doi.org/10.3389/fphar.2021.692574.
