PROTAC linkers have moved from being mere hydrocarbon straps to versatile multi-functional modules that can make or break a degrader in cells. Achieving the right linker properties in terms of length, rigidity, polarity and metabolic stability is now key to tuning ternary-complex geometry, cellular uptake and off-target liability—all while staying within a molecular weight budget that must remain low enough for oral exposure. The following sections catalogue the dominant structural classes, explain why the linker is now considered a pharmacological element in its own right, and outline practical considerations for sourcing or custom-building these critical fragments without the need of proprietary catalogues.
Introduction
The linker is the only region of a PROTAC that is simultaneously exposed to bulk solvent, to the ligase surface and to the target surface; as a consequence its chemical identity radiates through every downstream property. A well-chosen tether can rescue an otherwise mediocre warhead by pre-organizing the ternary complex, whereas a poorly matched spacer can sink a programme in spite of nanomolar binding. Understanding the available architectural palettes—flexible, rigid, heteroaryl, nitrogen-rich—therefore becomes as important as optimising the ligands themselves.
Why Linkers Are Critical in PROTACs?
In the cytosol, the linker serves as a transient molecular scaffold and must satisfy 3 (often competing) requirements. It must:
- (1) position the warhead and the E3 ligand in an orientation that enables cooperative protein–protein interactions (i.e., a single atom mismatch can convert positive cooperativity into a repulsive encounter, converting an otherwise effective degrader into a costly antagonist). It must
- (2) cross membranes: if too lipophilic, the hydrocarbon chain will bind albumin, if too polar, the PEG is sequestered in endosomes.
- (3) Finally, it must remain intact in the face of oxidative, hydrolytic and conjugative threats long enough to engage in multiple catalytic cycles (a single benzylic oxidation can reduce half-life from hours to minutes).
In view of these constraints acting in concert, the linker emerges as the integrative node at which medicinal chemistry, PK and structural biology converge. Iterative optimization therefore necessarily prioritizes the tether itself—tuning length, rigidity and polarity—before the warhead or ligase ligand is revisited. This hierarchy has now been codified in discovery workflows: early prototypes are intentionally designed around modular linkers that can be swapped in a single synthetic step, ensuring that the rest of the molecule is not overdesigned around an irreversible spacer choice.
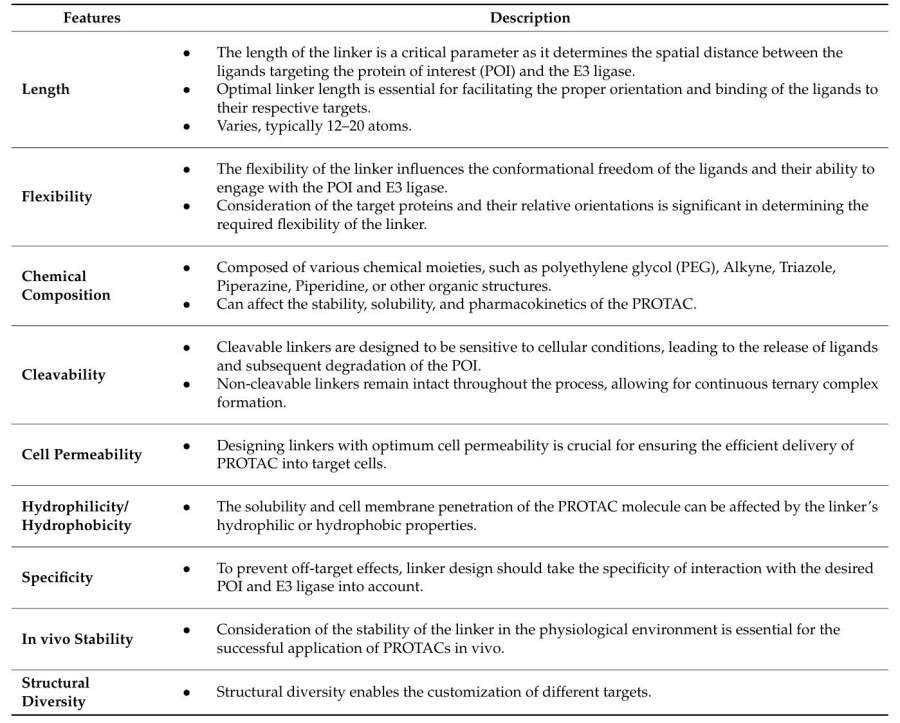 Fig. 1 Key features of linkers for PROTAC development.1,5
Fig. 1 Key features of linkers for PROTAC development.1,5
Overview of Linker Categories
Linkers are pragmatically classified by the dominant physical property they impart. Flexible families (linear alkanes, unsaturated alkenes, PEG chains) are frequently used for first-pass screening as they are one-step couplings from commercial diacids or diols, and their conformational generosity makes it more likely to hit any ternary complex, albeit at the expense of entropic penalties later. Rigid cohorts (triazoles, piperazines, cyclohexynes, para-phenylenes) lower rotational entropy, steepening dose–response curves and often improving oral exposure by reducing the number of rotatable bonds counted in absorption models. Heteroaryl fragments (pyridine, oxadiazole, thiazole) add directional dipoles that can recruit interfacial waters, effectively gluing the complex together without increasing molecular weight. The photoresponsive groups azobenzenes and nitrobenzyl esters allow for spatial and temporal regulation that restrains systemic activity to only the tumor bed exposed to fiber optic illumination. Macrocyclic and spiro variants provide binding site distances that are pre-organized to within one-tenth of a nanometer through their cavities which allow researchers to focus on one synthetically challenging scaffold for optimal geometry. Each category is accompanied by off-the-shelf building blocks sporting orthogonal protecting groups, allowing investigators to order milligrams for biochemical proof-of-concept or grams for in vivo studies without revisiting route scouting.
Types of PROTAC Linkers
PROTAC linkers are usually classified into 4 architectural families: flexible PEG, hydrophobic alkyl, planar aromatic and heteroaryl/nitrogen-rich. These classes encode a unique combination of entropy, polarity and metabolic fate. Selection between them is rarely an either/or decision and many successful degraders are hybrids that combine motifs (e.g., alkyl–triazole or PEG–piperidine) to fine-tune length, rigidity and solubility within a single spacer. The sections below summarise the signature traits of each class, and highlight why a hybrid strategy is now the default rather than the exception.
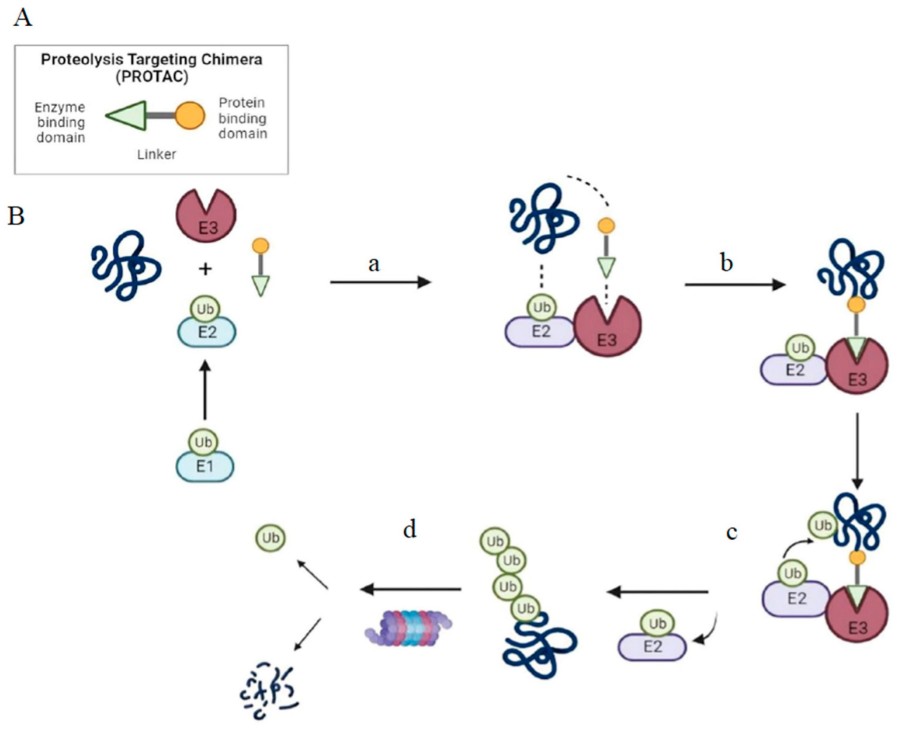 Fig. 2 Structure and mechanism of PROTAC-based degradation.2,5
Fig. 2 Structure and mechanism of PROTAC-based degradation.2,5
PEG oligomers remain the default choice as a hydrophilic tether, in part because their structural code readily encodes two properties: conformational flexibility and water solubility, both in a chemically transparent manner. The repeating ether oxygen atoms increase polar surface area without adding charges, allowing the PROTAC to remain un-ionized yet easily dispersible in aqueous solutions. In terms of conformation, PEG is like a molecular spring: it adopts an ensemble of extended and collapsed states which increases the likelihood that the warhead and ligase ligand simultaneously find their respective pockets during a single diffusional encounter. This conformational adaptability is valued, in particular, in early-stage campaigns where the optimal separation distance is not yet known; by simply adding or subtracting one ethylene glycol unit, one gains a 3-bond (≈3.6 Å) length increment without redesigning the synthetic route. The ether backbone is also more resistant to oxidative backbone cleavage than polymethylene, but is not metabolically inert: sequential O-dealkylation is possible, particularly when the chain is greater than six units, which would result in shorter, more lipophilic fragments that would complicate cellular read-outs. A second shortcoming of PEG is molecular-weight inflation: when the PEG segment becomes too long, it dominates the mass profile and takes the construct outside of the oral exposure window. For this reason, contemporary practise often limits PEG to short oligomers (n = 2–6) or pairs it with rigid heteroaryl caps that restore the aspect ratio without adding glycol mass. Despite these shortcomings, PEG remains the default hydrophilic module for injectable or early-cell-assay PROTACs because its physicochemical signature is predictable and the chemistry is trivial: any terminal azide, amine or alcohol can be extended overnight under aqueous conditions.
Straight-chain alkyl tethers persist in orally active degraders, as they provide hydrophobic span with low atomic overhead. The repeating methylene unit encodes exactly 2.5 Å of distance, so length can be scanned with experimental precision without the need for computational modelling. In contrast to PEG, alkyl chains add no polar surface area, so they compress logP and molecular weight while maintaining membrane permeability—a trade-off which often results in improved oral exposure. Flexibility is both an asset and a liability: the chain can collapse into small, hair-pin conformations, which diminish the entropic penalty of ternary complex assembly, but the same mobility increases the likelihood of metabolic attack. Cytochrome P450 enzymes abstract benzylic or allylic hydrogens, to form alcohols which can be further oxidised to carboxylic acids or ketones; these metabolites are often more polar, and may be retained, which confounds interpretation of in-vivo studies. To avoid this liability, modern alkyl linkers are terminated by electron-withdrawing amides or capped with short rigid motifs (alkyne, cyclopropane) that prevent ω-oxidation while maintaining the low-mass advantage. Branching is generally avoided, as it increases logP disproportionately, but a single methyl group placed γ to the nitrogen can protect the adjacent C–H bond with little impact to aspect ratio. Overall, short alkyl segments (C4–C10) remain the permeability module of choice when the therapeutic index is limited by exposure rather than potency.
Aromatic rings transform the linker from a passive strap into a conformationally locked rod, the length of which is set by π-bond geometry instead of rotatable bonds. Phenyl, biphenyl or phenyl-ethynyl units enforce a planar, hydrophobic spine that pre-organises the PROTAC into an extended conformation and steepen the degradation curve by lowering the entropic cost of ternary complex assembly. The delocalised π-cloud also engages in edge-to-face or offset stacking, and the phenylalanine, tyrosine or histidine side chains that frequently line the target–ligase interface can provide enthalpic glue to supplement the binary affinities of the terminal ligands. Metabolically, the electron-rich ring is less susceptible to oxidative cleavage than alkyl chains, hydroxylation occurs preferentially at para positions and can be blocked by small fluorine substituents without perturbing π-stacking. The principal downside is solubility: sequential aryl rings increase crystal lattice energy and can trigger colloidal aggregation in the micromolar range. This limitation is usually circumvented by inserting a single heteroatom (ether, sulfone) or capping the aromatic stack with a basic nitrogen, strategies that disrupt symmetrical packing but maintain rigidity. Synthetic accessibility is no longer an issue: brominated or boronated aromatics are commodity reagents that couple under aqueous Suzuki conditions, allowing rigidification to be explored in parallel rather than in series.
Heteroaryl linkers incorporate aspects of both aromatic (rigidity) and aliphatic (polarity, hydrogen-bond directionality from N lone pair) motifs. Pyridine, pyrimidine, triazole or imidazole, for instance, provide an sp² nitrogen whose lone pair can act as a hydrogen bond acceptor for backbone NH groups to help staple the developing ternary complex without contributing to MW. Regiochemistry can be another dial: 3-pyridyl, for example, buries the lone pair, while 4-pyridyl aligns the dipole moment vector with the linker axis, which may be desirable if the interface also requires an electrostatic zipper motif. Saturated nitrogen rings (piperazine, morpholine, azepane) can provide pH-tunable basicity to enhance endosomal escape, but are largely neutral at cytosolic pH, thus more amenable to good oral exposure without permanent charge. Heteroaryl rings also tend to be metabolically more stable than phenyl: the electron-deficient ring is less prone to oxidative metabolism, and when that occurs N-oxidation is preferred, a pathway that generally results in polar, excretable metabolites. Synthetic orthogonality with azides, boronic acids and halogenated heteroarenes is also high, since those units couple under mild and functional-group-tolerant conditions, enabling rapid hit-to-lead iteration which is more difficult to achieve when working with heavily substituted alkyl chains. Together, heteroaryl and nitrogen-based motifs can be used as multi-tasking modules to encode rigidity, polarity, and recognition cues within a single, lightweight unit, and are currently the fastest-growing linker category in PROTAC research.
Applications in Drug Discovery
PROTAC linkers have evolved from inert tethers to pharmacologically functional modules that define both efficacy and developability across therapeutic areas. In oncology, rigid heteroaryl spacers have been employed to enable oral degradation of transcription factors long considered undruggable, while in neurodegeneration, nitrogen-rich flexible linkers have been leveraged to enable blood–brain-barrier penetration and disaggregation of misfolded proteins. Here, we highlight several case studies to show how relatively minor linker edits (linker length, rigidity or basicity) can be translated into quantitative therapeutic gains without modification of the terminal ligands.
Oncology Programs
Recent case studies from oncology drug discovery demonstrate that linker choice can mean the difference between a PROTAC that languishes as a laboratory curiosity or one that quickly enters drug-like chemical space. In one such example, PROTACs against an oncogenic transcription factor were improved by swapping a flexible alkyl tether for a short, planar, nitrogen-rich rod. A pyridine–triazole hybrid linker dipole was oriented against a backbone carbonyl at the target–ligase interface. Structural studies showed that a network of water-mediated polar interactions anchored by the triazole nitrogen formed at the interface. In contrast, this contact was not observed for the alkyl parent. Wash-out assays in cells suggested the improvement in tumour-growth inhibition at nanomolar exposure was a result of slower dissociation, rather than an increase in the binary affinity. In a second case study, a piperazine-capped PEG linker was used to degrade a DNA-repair helicase. Basic nitrogen improved solubility to the point that oral delivery became possible. The linker PEG segment also provided sufficient flexibility to bind the shallow, solvent-exposed groove of the helicase. Rodent studies showed tumor regression with no measurable bone-marrow toxicity. This was attributed to the linker's ability to sterically restrict ternary-complex formation to tumor cells over-expressing the ligase adaptor. The unifying feature of these two apparently divergent targets is that linker edits were not merely cosmetic, but remedied specific solubility, orientation and metabolic liabilities that had tripped up earlier analogues. This is why linker optimization is now routinely done in parallel with ligand optimization.
Neurodegenerative Research
The special requirements of neurodegeneration – blood–brain-barrier penetration, low neuronal proliferation and the need to disaggregate misfolded proteins – may make linker design even more important than in oncology. In one recent example, a C-terminal fragment of TDP-43, a pathological species in amyotrophic lateral sclerosis, was chosen as a target. A neutral, tetra-ethylene-glycol linker was used to trade some polarity for flexibility, allowing the PROTAC to pass through the narrow synaptic cleft without permanent charge. Advanced imaging showed that the compound reduced aggregate load and oligomeric compactness in neuronal cultures, and increased motility in transgenic nematodes, without affecting endogenous full-length TDP-43. In another tau-directed programme, a rigid biphenyl rod was replaced by a morpholine–urea hybrid. The morpholine oxygen functioned as an acceptor of a hydrogen bond from a backbone NH on tau's microtubule-binding repeat, while two urea NH donors interacted with a pair of carbonyls on the recruited E3 ligase, forming a transient but directional staple. Brain exposure doubled relative to the biphenyl parent and memory deficits in a murine model were reversed after a short oral regimen, exemplifying how nitrogen-rich, moderately flexible linkers can mediate CNS permeability and ternary-complex stability. These cases exemplify how successful neurodegeneration PROTACs often trade extended aromatic rigidity for shorter, polarity-tuned spacers that can navigate the extracellular matrix, cross endothelial tight junctions and still direct the orientation of the pathogenic protein for ubiquitination inside post-mitotic neurons.
Ordering and Sourcing Guide
At BOC Sciences, we simplify the process of sourcing high-quality PROTAC linkers for research and development. Whether you need PEG, alkyl, aromatic, or heteroaryl linkers, we provide a scalable, reliable supply chain and expert consultation to ensure every compound meets your project's specifications.
Stock Options and MOQ
We maintain a diverse in-stock portfolio of PROTAC linkers covering all major categories:
- PEG linkers (PEG2-PEG8) for flexibility and solubility.
- Alkyl linkers (C3-C12) for membrane permeability and lipophilicity.
- Aromatic and heteroaryl linkers for rigidity and π–π interactions.
- Functionalized variants including amine, azide, alkyne, and NHS handles for easy conjugation.
Our minimum order quantity (MOQ) starts from 100 mg for early-stage screening, making it ideal for both academic labs and biotech R&D programs. Each batch is accompanied by a Certificate of Analysis (COA) and analytical data (HPLC, LC-MS, NMR) to confirm purity and consistency. Standard products are available for global shipment within 3-5 business days, with temperature-controlled logistics to ensure product integrity.
Bulk Orders and Custom Synthesis
For larger-scale or specialized research programs, we offer bulk manufacturing and custom synthesis services to meet your exact requirements. Our capabilities include:
- Multi-kilogram production for advanced development and preclinical studies.
- Custom linker design, allowing modifications to chain length, polarity, or functional groups.
- Hybrid linker synthesis (PEG-alkyl, PEG-aromatic, or PEG-heteroaryl combinations).
- Isotopic labeling and GMP-adjacent production for regulated environments.
Our chemists collaborate directly with your R&D team to ensure chemical compatibility with E3 ligases (VHL, CRBN, MDM2, IAP) and target proteins. We guarantee transparent lead times, competitive pricing, and full analytical documentation with every delivery.
Partner with Us for Comprehensive PROTAC Linker Solutions
As the field of targeted protein degradation evolves, the choice of linker remains a decisive factor in PROTAC success. The right linker-balanced in length, polarity, and rigidity-can define a molecule's potency, selectivity, and bioavailability. At BOC Sciences, we deliver end-to-end PROTAC linker solutions, from stocked PEG and alkyl linkers to fully custom-designed aromatic and heteroaryl scaffolds. Our team combines chemistry expertise, fast global logistics, and responsive support to accelerate your discovery pipeline.
Contact our technical experts today to request a quote, browse available linkers, or initiate a custom synthesis project tailored to your program. Empower your next-generation PROTAC research with linkers engineered for performance, precision, and scalability.
High-Purity PROTAC Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity linkers optimized for targeted protein degradation.
FAQs
1. What types of linkers are used in PROTACs?
Common types include PEG (flexible), alkyl (permeable), aromatic (rigid), and heteroaryl (directional) linkers.
2. How do I choose the right linker type?
Base your choice on your target–E3 pairing, required solubility, and ternary complex stability.
3. Do you offer bulk or custom synthesis?
Yes, we supply all linker classes in research and bulk quantities with full analytical validation.
4. Can I get expert help selecting linkers?
Yes, our team offers free consultation to help you identify the most suitable linker for your program.
References
- Danishuddin, Jamal M S, Song K S, et al. Revolutionizing drug targeting strategies: integrating artificial intelligence and structure-based methods in PROTAC development[J]. Pharmaceuticals, 2023, 16(12): 1649. https://doi.org/10.3390/ph16121649.
- Ahmad H, Zia B, Husain H, et al. Recent advances in PROTAC-based antiviral strategies[J]. Vaccines, 2023, 11(2): 270. https://doi.org/10.3390/vaccines11020270.
- Arthur R, Valle-Argos B, Steele A J, et al. Development of PROTACs to address clinical limitations associated with BTK-targeted kinase inhibitors[J]. Exploration of targeted anti-tumor therapy, 2020, 1(3): 131. https://doi.org/10.37349/etat.2020.00009.
- Zagidullin A, Milyukov V, Rizvanov A, et al. Novel approaches for the rational design of PROTAC linkers[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 381. https://doi.org/10.37349/etat.2020.00023.
- Distributed under Open Access license CC BY 4.0, without modification.
