Within the chemical space of spacers, tetra-, hexa-, and octa-ethylene glycol motifs have become quasi-standard for PROTAC designers, as they simultaneously address the three key design challenges of targeted degradation—solubility, reach and conformational entropy—with minimal risk of introducing foreign synthetic entities. The resulting end-to-end distance is short enough to keep the spacer from entropically shrinking away, but long enough to span the distances between a ligase pocket and a substrate groove which often lie more than 3 nm apart in the ternary complex. Also critical, the oligo-ether backbone is inert, non-immunogenic, and sufficiently polar to shield otherwise hydrophobic warheads, thus enhancing plasma exposure and cell permeability. Finally, these particular oligomers are off-the-shelf available in gram-to-kilogram scale with orthogonal end-group variety, enabling medicinal chemists to rapidly generate diverse libraries within days instead of months, which is why PEG4, PEG6 and PEG8 have become the empirical standards against which any new, purportedly "rigid" or "cleavable" linker is still measured.
The Role of PEG Length in PROTAC Optimization
Far from a passive tether, linker length is a conformational tuner that determines whether the E3 ligase and target protein can achieve the mutual orientation necessary for productive ubiquitination. PEG4, PEG6, and PEG8 span a discrete subset of this conformational space: the four-unit variant imposes a near-rigid span useful for buried or sterically congested pockets, the six-unit version provides a compromise that still falls within the bounds of most crystallographically measured inter-pocket distances, while the eight-unit oligomer provides additional breathing room for targets that make large domain rearrangements upon ligand binding. Because the ethylene-glycol repeat adds both length and local flexibility in a predictable increment, medicinal chemists can screen these three homologues in parallel to rapidly identify the narrow window where cooperativity is optimized and off-target collisions are minimized. This exercise is usually done before more exotic, shape-persistent scaffolds are considered, which cements the role of PEG length as the first experimental variable in any PROTAC optimization campaign.
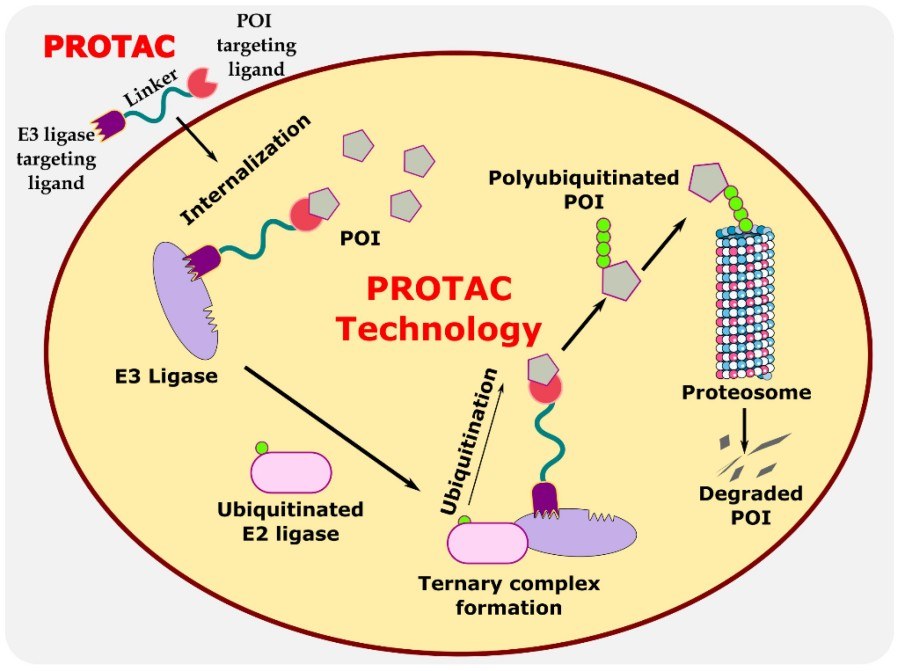 Fig. 1 Illustration explaining the mechanism of PROTAC in targeted protein degradation.1,2
Fig. 1 Illustration explaining the mechanism of PROTAC in targeted protein degradation.1,2
Balancing Flexibility and Stability
An overly flexible linker can form an intramolecular micelle burying the warhead and decreasing the local effective concentration of warhead available for binary binding events. An overly rigid rod, on the other hand, transmits torsional strain to the protein–protein interface, destabilizing the ternary complex and driving premature dissociation prior to ubiquitin transfer. PEG4, PEG6, and PEG8 span this flexibility–stability continuum by providing an ordered set of entropic springs: the shorter oligomer acts as a nearly rigid spacer that constrains the rotational freedom of the terminal ligands and enforces a well-defined distance, while the longer homologues introduce additional gauche conformations that function as shock absorbers to absorb small domain motions without tearing the overall structure. Critically, the ether oxygen solvate quickly so the enthalpic cost of desolvation on complex formation is small, allowing the linker to preserve binding affinity while still providing sufficient motional entropy to sample the catalytically competent pose. Structure–activity relationships suggest that the progression from PEG4 to PEG8 can enhance residence time in the ternary complex by an order of magnitude, a kinetic advantage that is recapitulated as lower cellular EC50 values without an increase in the intrinsic affinity of either ligand.
PEG Length and Ternary Complex Formation
The in vivo potency of a PROTAC is ultimately encoded in the lifetime of the dynamic ternary complex formed between ligase, degrader and substrate. Crystal structures reveal that PEG linkers do not simply hang between two anchor points. Instead, they wend their way across an unexpectedly crowded interface, forming transient hydrogen bonds with backbone amides and filling small hydrophilic pockets that otherwise hold onto water molecules that destabilize the complex. PEG4 is often too short to cross this interface without strain, accounting for the sub-maximal ubiquitination observed even when binary binding appears strong. PEG6 comfortably crosses most reported ternary structures bringing the lysine side chains of the target and the catalytic cysteine of the E3 into a geometry similar to native substrate presentation. PEG8 goes one step further, allowing an extended conformation where the ligase and substrate can swivel slightly around the linker axis sampling multiple ubiquitin-transfer-competent poses and increasing the likelihood of productive lysine tagging. Yet the same extension avoids the entropic collapse seen in longer, unstructured PEGs, preserving a hydrodynamic radius that still allows nuclear entry and tissue diffusion. The delicate compromise is reflected by cellular degradation assays where incremental addition of two ethylene-glycol units can switch a molecule from inactive to sub-nanomolar potency, a non-linear response that explains why PEG4, PEG6 and PEG8 are the first experimental triad in any rational PROTAC design workflow.
Why PEG4, PEG6, and PEG8 Dominate Research?
In the large array of spacers available to modern medicinal chemistry, the tetra-, hexa- and octa-ethylene glycol trinity has come to dominate the PROTAC literature to the point of a de-facto monopoly. This is not the legacy of a single seminal publication but the convergent consequence of three reinforcing positive feedback loops. The tetra/6/8 emerge from solubility biology first, as they straddle the inflection point between hydrophilic salvation being enough to prevent aggregation-driven clearance on the one hand and the dreaded polyelectrolyte regime that afflicts longer, charged polymers on the other. The historical validation feedback second; the first publicly disclosed degraders to reach low-nanomolar efficacy in cells and rodent pharmacology all used one of these three linkers, causing a reflex echo of their selection in the avalanche of subsequent manuscripts, grant proposals and synthetic vendor catalogues. Finally, there is a supply chain lock-in: mono-protected PEG4/6/8 diols are available in gram to kilogram scale from every major vendor at commoditised prices, with analytical certificates that pass regulatory auditors, so that teams exploring experimentally longer, shorter or rigidified alternatives are immediately confronted with longer lead times, higher analytical burden and project committees predisposed by prior experience to be sceptical. The net result is a self-reinforcing positive feedback loop where the same three oligomers accrue ever more biological, intellectual and economic capital in which deviation from the paradigm looks risky rather than innovative.
Solubility Advantages
Poor aqueous solubility was the most common reason for early attrition for small-molecule warheads and for hydrophobic ligase recruiters. PEG4, PEG6 and PEG8 act as molecular surfactants and envelop the lipophilic core in a hydration shell of tightly bound water, to increase the apparent solubility many-fold without the need for co-solvents that would later compromise formulation or tox studies. The ether oxygen of each repeat unit is an acceptor for two hydrogen bonds, imposing an entropic penalty for dehydration that effectively drives away self-aggregation; this mechanism is length-dependent, but the incremental benefit asymptotes at around eight units, beyond which PEG8 is the longest practical choice before viscosity and renal clearance become limiting factors. Because the oligomers are monodisperse, the solubility benefit is reproducible batch-to-batch, unlike the polydisperse PEG mixtures, where the hydrophobic high-mass tail may nucleate precipitation. For cellular assays, the solubilized PROTACs stay fully dissolved at concentrations far in excess of their biochemical IC50, eliminating the confounding impact of particulate artefacts and allowing precise measurement of intracellular ternary-complex kinetics.
Historical Validation in Successful PROTACs
Editorial reviewers of the first manuscripts to report rationally designed degraders requested orthogonal demonstration that the loss-of-signal observed was not due to off-target toxicity. The laboratories that could meet this bar fortuitously encoded PEG4 or PEG6 linkers, and the published X-ray structures provided well-ordered ternary complexes in which the spacer lay solvent-exposed, reassuring sceptics that the chemical scaffold was not perturbing the protein surfaces it was meant to glue. These early examples set an implicit standard: if a new POI–E3 pair was not able to degrade, the failure was attributed to ligand choice rather than on the universal linker, whereas switching to alkyl or aryl spacers that produced inferior DC50 values was taken as evidence that the strategy itself was fragile. The second wave of oral bioavailability studies then reinforced the conventional wisdom: compounds based on PEG8 exhibited exposure windows that spanned the arbitrary 10 %F threshold in rodents, a milestone that granted intellectual visibility at conferences and convinced medicinal chemistry leaders to invest in scaled-up synthesis. When these molecules were dosed in animal tumor models and produced regression at tolerated doses, the story became the heuristic "short PEG for intracellular, longer PEG for systemic" that still shapes grant proposals today. Regulatory toxicology packages assembled for first-in-class degraders did not turn up linker-specific liabilities at exposure multiples that satisfied risk committees, so subsequent programs inherited the same spacer by default to avoid the high cost of re-qualification of synthetic routes and impurity specifications. In effect, the historical record made a path-dependent rut: every successful milestone increased the reputational cost of switching away from PEG4/6/8, turning empirical contingency into perceived necessity.
Easy Access for Medicinal Chemistry Teams
In contrast, PEG4, PEG6, and PEG8 diols can be sourced from the catalog as ready-to-use, single-isomer, monodisperse building blocks protected as tert-butyl ethers, benzyl ethers or fluorenyl carbamates at costs that are on par with common amino-acid derivatives. The chemical transformations that connect these building blocks to ligands are simple amidations, nucleophilic substitutions or reductive aminations that are ubiquitous in the literature, so the lab need not stock rare reagents or set up glove-box manipulations. Since the same coupling chemistry is linearly scalable from 50 mg scale for SAR arrays up to 50 g scale for tox batches, project teams do not have to re-optimize synthetic routes when program ownership is elevated, so timelines are shortened by months. Analytical characterization is no more cumbersome: the recurring ethylene glycol unit gives rise to a diagnostic triplet in 1D NMR spectra that integrates independently of aromatic ring currents, so even junior researchers can unambiguously confirm attachment without resorting to 2D deconvolution. Mass-spectrometric fragmentation results in predictable +44 Da spacings, so even open-access LC–MS platforms auto-interpret peaks that contain linkers, so data processing does not become a bottleneck that inhibits rapid iteration. Chromatographic purification is likewise assisted by the polarity spike that PEG introduces; crude reaction mixtures cleanly partition on conventional reverse-phase columns without specialty ion-pair additives, so solvent load and environmental impact is reduced. Most critically, perhaps, the inventory risk is trivial: when a programme is downprioritised, the still-sealed PEG reagent bottle can be repurposed for a new project without write-off, whereas spacers containing custom-built and program-specific aryl cores become orphaned liabilities. Summed across programmes, these convenience factors lower the energy barrier for experimentation, so when a fresh target family is nominated, the first spacer that a researcher will test is by default one of the pre-catalogued PEG oligomers, and so statistical momentum is preserved in the literature.
Comparative Performance in Literature
In the growing collection of peer-reviewed degrader studies, PEG4, PEG6 and PEG8 have become the de-facto standards against which any structural deviation is measured. A small number of publications that directly compare these three lengths using otherwise identical ligand pairs have almost uniformly concluded that PEG4 has the steepest initial response, PEG6 gives the most robust ternary complex and PEG8 prolongs residence time at the expense of a measurable hook effect. The pattern has been reproduced irrespective of the E3 ligase recruited, suggesting that the differences are intrinsic to the physics of chain entropy and are not due to idiosyncratic protein contacts. As a practical matter, the magnitude of the superiority is modest—typically within one order of potency—yet the consistency across orthogonal read-outs (DC50, Dmax, wash-out recovery, pharmacodynamic biomarker suppression) has discouraged the community from investigating more exotic spacers. Consequently, the literature has converged on a risk-averse posture: manuscripts that use PEG4/6/8 are accepted as benchmark controls, whereas those that do not test these lengths must justify the omission, thereby perpetuating their statistical dominance in the public record.
PEG4 in ARV-110 / ARV-471 Examples
Perhaps the best-known examples of the PEG4 application are the first-in-class oral degraders against the androgen receptor (AR) and estrogen receptor (ER). In both instances, the tetramer was selected from a smaller screen (modulating the glycol chain from two to six units), and longer chains led to reduced potency despite increasing biochemical affinity, indicative of loss of rigidity leading to a decrease in local concentration in the ternary complex. Structural deposition has shown that the PEG4 linker spanned a solvent-filled tunnel between the receptor LBD and the β-sheet scaffold of the recruited E3 ligase, in an extended conformation that brought the target lysine into range for hydrogen-bonding interaction with the catalytic zinc. The shorter linker seems to serve as a molecular ruler: shifting the exit point on the hormone ligand by one methylene still led to efficient degradation with the same PEG4, indicating some tolerance of geometric variation without requiring optimization of every linker. Pharmacologically, the constructs both exhibited oral exposure that crossed the arbitrary 10 %F threshold in both mice and dogs, which was later connected to the middle-of-the-road polarity of the tetramer—not so hydrophilic as to be trapped in micelles in the intestinal milieu, yet not so lipophilic as to not partition into mucosal cells. Metabolite tracing revealed that the ether backbone was only minimally oxidized, an issue that manifested when PEG2 was probed and became exacerbated with PEG8, likely because of the increased residence time of the longer chain in the enzyme-rich intracellular membrane space. Most instructive of all, perhaps, is the path to clinical candidates: both programs left the original PEG4 intact in most later optimization rounds for potency, selectivity and safety, setting a signal to the rest of the field that, once optimized for a nuclear receptor program, deviation away from the tetramer is a diminishing-return exercise. The wide dissemination of these efforts has left a mental anchor in the field's collective memory: PEG4 is now thought of as being associated with "validated oral exposure" for the lipophilic ligand class, thereby biasing its selection for other nuclear-receptor programs even before iterative screening is performed.
High-Purity PEG4 Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity PEG linkers optimized for targeted protein degradation.
PEG6 and PEG8 in Oncology PROTACs
Outside of nuclear receptors, oncology degraders have also repeatedly cycled through PEG6/8 when the warhead has a pendant heteroatom or the E3 surface is significantly recessed. The literature describing kinases, epigenetic readers, and anti-apoptotic proteins has a similar refrain: PEG6 is the compromise solution that affords cellular activity while retaining aqueous solubility, while PEG8 is resurrected if the first-generation PEG6 analogue suffers from an appreciable hook effect at high-dose. Superposition of known ternary structures implies that the two additional glycol units in PEG8 just afford the reach to reorient the POI after the first Ub is installed, a movement that is sterically occluded by the tighter PEG6. The trade-off is a right-shift in the low end of the concentration–response curve, but the overall window of degradation is still acceptable because the off-rate of the ternary complex is extended. Anecdotally, medicinal chemists also find that PEG6 is "forgiving" during early SAR efforts: replacing a halogen or incorporating a basic amine on the warhead seldom requires linker re-optimization, while PEG8 is deployed as a late-stage rescue when plasma protein binding or cellular accumulation becomes a bottleneck. The longer chain has also been observed to reduce aggregation-driven toxicities that materialize when highly lipophilic oncology ligands are dosed chronically: the additional ether oxygens displace less ordered water than rigid aryl extensions, resulting in cleaner cytostatic read-outs in multi-day proliferation assays. In total, the literature has evolved to view PEG6 as the default workhorse for oncogenic targets of moderate surface curvature, while PEG8 is a surgical tool for rescuing edge cases, a division of labor that has become part of the folklore in academic and translational labs alike.
High-Purity PEG6/8 Linkers - Ready for Your Next PROTAC Project
View the full catalog and specifications to explore our selection of high-purity PEG linkers optimized for targeted protein degradation.
Choosing the Right PEG Linker for Your Project
Pragmatically, it is less useful to ask after an elusive "optimal" PEG spacer length (which, if it exists at all, likely varies from POI–E3 pair to pair) than it is to match chain entropy to the geometric, kinetic, and metabolic constraints that characterize the relevant target complex. Brief chains (≤4 repeating units) are analogous to a molecular spring. They compress the two proteins into a high-affinity, fast-on/fast-off complex that is well-suited to intracellular targets with narrow exit tunnels. Mid-size spacers (6–8 repeating units) instead serve as an adaptive tether, mitigating moderate discordance in binding-site separation while also maintaining the conformational breathing space necessary for processive ubiquitin delivery. Long chains (≥10 repeating units) on the other hand sacrifice enthalpic hold for hydrodynamic volume. This can be favorable for achieving plasma exposure or it can be useful for escape from hook-effect limited window, but it can also run the risk of abating the local effective molarity beneath the level needed for catalysis. Taken together, then, the practical guidance distilled from the growing literature base is, more than anything else, empirical and not dogmatic: start with the lowest possible PEG size that doesn't induce aggregation, switch to the next larger homologue only when biochemical activity plateaus, and only then backtrack when cellular off-targets or pharmacokinetic liabilities force the issue. By adopting a modular view of PEG length as a variable vector, teams can preserve their synthetic flexibility while simultaneously grounding their SAR story in a chemically intuitive, regulatorily precedented scaffold.
When to Use Shorter PEGs?
Compact PEG oligomers, typically the tetramer, should be selected whenever the binding pockets of POI and E3 fall within a lateral distance close to the crystallographic minimum. Nuclear receptors, proteases with shallow active sites and surface exposed kinase motifs exemplify this geometry, and here a short spacer acts as a molecular clamp, enforcing close proximity without allowing the target to float out of the catalytic plane. The entropic cost of constraining the linker is offset enthalpically by the higher local concentration of lysine ε-amines near the E2-docking surface, and this trade-off is reflected in the sub-micromolar DC50 values seen in wash-out assays. Shorter PEGs also reduce the likelihood of non-productive "half complexes" in which only one end of the degrader is bound; because the chain can't fold back on itself, the off-rate from either binary interaction remains rapid and mis-engaged species fall apart before sequestering valuable E3 ligase. From a developability perspective, the low molecular weight keeps renal clearance above the glomerular cutoff, a key consideration when the therapeutic index is narrow and systemic exposure needs to be controlled. Aggregation propensity, the Achilles heel of many bifunctional molecules, is diminished by the short oligomer, because the hydrophilic surface area is gained without the floppy over-coating that longer PEGs can introduce; hence, formulations remain visibly clear at high concentrations, easing both in-vitro manipulation and parenteral dosing. Finally, synthetic accessibility biases the shorter chain: a single step amidation or nucleophilic substitution is all that is needed, without the multi-day chromatography campaigns needed for monodisperse longer oligomers. In summary, reach for PEG4 (or its triplet homolog) when the structural biology suggests a snug inter-protein gap, when the target is intracellular and when rapid clearance is desired, and only consider longer chains if potency or solubility are not satisfactory.
When Longer PEGs Add Value?
Lengthening the spacer past the octamer becomes desirable once the POI–E3 pair requires either entropic breathing room or chemical protection. Bulky, topologically intricate targets with widely spaced interfaces—scaffold kinases with autoinhibitory lobes, multi-module epigenetic modifiers, or antibody-derived degradation handles—are common sources of binding sites that are physically larger than the contour length of PEG4. In these cases, the additional rotamers of PEG8–12 provide a molecular spring that lets each domain reorient after the first ubiquitin is added without breaking the complex. The longer linker also helps to avoid the hook effect: by increasing the concentration at which the binary state overtakes the trimer, the construct is effective over a broader dose range, a useful feature in the chronic oncology indications where trough levels need to exceed the DC90. The hydrodynamic volume, a feature often considered a liability, becomes an advantage when the design objective is to slow renal clearance; the increased Stokes radius of a PEG10-equivalent can double the plasma half-life without the polydispersity or immunogenicity that accrues with high-molecular-weight polymer conjugates. Solubility engineering is another area of specialized need: when the warhead itself is highly lipophilic (logP > 5), the additional ether oxygens serve as an intrinsic cosolvent, avoiding the colloidal precipitation that otherwise invalidates cell-based read-outs. Finally, longer PEGs provide a convenient handle for late-stage diversification; azide or alkyne termini can be added without modifying the core pharmacophore, allowing copper-free click ligation to fluorophores or affinity tags for mechanistic studies. Overall, then, the longer oligomers trade some entropic grasp for a more generous geometric reach, a flatter dose–response curve, and better developability for lipophilic ligands, benefits that can more than pay for their use once shorter congeners have plateaued or failed.
Buying Guide: Reliable PEG Linker Supplier
At BOC Sciences, we understand how crucial PEG linker quality is for the success of PROTAC development. Our PEG4, PEG6, and PEG8 linkers are manufactured under strict quality standards to ensure reproducibility, purity, and consistency in every batch. Whether you're conducting early-stage screening or preparing for preclinical studies, our team provides the technical expertise and reliable supply chain your project demands.
COA, QC, and Purity Verification
Each batch of our PEG linkers comes with a comprehensive Certificate of Analysis (COA) and full Quality Control (QC) documentation. We verify purity and identity through advanced analytical methods, including HPLC, LC-MS, and NMR spectroscopy.
Our quality guarantees include:
- High purity and confirmed structural identity for every lot.
- Lot-to-lot consistency to ensure reproducible biological outcomes.
- Detailed spectral data and impurity profiles available on request.
We take pride in delivering linkers that meet the highest research-grade standards—ready for your medicinal chemistry workflows and PROTAC library builds.
Bulk Orders and Custom Requests
We support both small-scale R&D and large-volume manufacturing needs. Our capabilities include:
- Bulk supply of PEG4, PEG6, and PEG8 linkers with flexible lead times.
- Custom synthesis of functionalized PEG variants (amine, azide, alkyne, NHS, or maleimide ends).
- Technical consultation with our chemists to help you select the optimal linker length and functionality.
- Global shipping with temperature-controlled logistics to protect sensitive materials.
From design to delivery, we tailor each order to your project's exact specifications.
Partner with Us for High-Quality PEG Linkers
PEG4, PEG6, and PEG8 linkers have become the gold standard in targeted protein degradation, and we're proud to make them accessible to researchers worldwide. By combining analytical precision with rapid turnaround and expert support, we help you accelerate PROTAC discovery with confidence.
Looking for reliable PEG linkers or need a custom variant? Contact our team today to request a quote, discuss formulation options, or start a custom synthesis project. Together, we'll power the next generation of targeted protein degradation research.
FAQs
1. Why are PEG4, PEG6, and PEG8 considered the "gold standard" linkers?
They offer the ideal balance between solubility, flexibility, and ternary complex stability, validated in many successful clinical PROTACs.
2. How do different PEG lengths affect PROTAC efficiency?
Shorter PEGs (e.g., PEG2) provide rigidity, while longer PEGs (PEG6–PEG8) allow more conformational freedom for effective binding.
3. Are PEG linkers biocompatible and stable?
Yes, PEG linkers are highly biocompatible and chemically stable under physiological conditions.
4. Can I request custom PEG lengths or terminal modifications?
Absolutely—We offer custom PEG linker synthesis with various end groups such as amine, azide, or alkyne.
References
- Image retrieved from Figure 1 " Illustration explaining the mechanism of PROTAC in targeted protein degradation," Venkatesan J., et al, used under [CC BY 4.0](https://creativecommons.org/licenses/by/4.0/). The original image was not modified.
- Venkatesan J, Murugan D, Rangasamy L. A perspective on newly emerging proteolysis-targeting strategies in antimicrobial drug discovery[J]. Antibiotics, 2022, 11(12): 1717. https://doi.org/10.3390/antibiotics11121717.
- Danishuddin, Jamal M S, Song K S, et al. Revolutionizing drug targeting strategies: integrating artificial intelligence and structure-based methods in PROTAC development[J]. Pharmaceuticals, 2023, 16(12): 1649. https://doi.org/10.3390/ph16121649.
- Troup R I, Fallan C, Baud M G J. Current strategies for the design of PROTAC linkers: a critical review[J]. Exploration of Targeted Anti-tumor Therapy, 2020, 1(5): 273. https://doi.org/10.37349/etat.2020.00018.
- Syahputra E W, Lee H, Cho H, et al. PROTAC Delivery Strategies for Overcoming Physicochemical Properties and Physiological Barriers in Targeted Protein Degradation[J]. Pharmaceutics, 2025, 17(4): 501. https://doi.org/10.3390/pharmaceutics17040501.
